2022
10-01
10-01
叙说“热图”
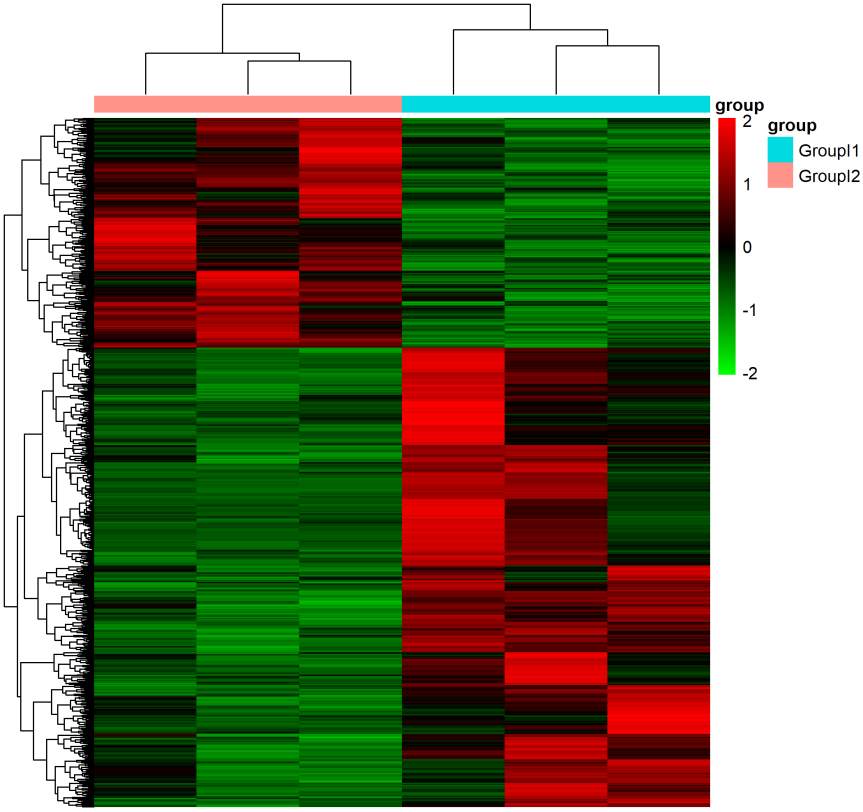 原理提起热图,大家可能马上就能领会:热图不就是差异表达基因在不同分组样品中的表达水平吗?一般一列表示一个样品,一行表示一个基因,其中的颜色表示的相对表达值。但说到热图与机器学习,很多人都会惊讶:一个简单的热图与高大上的机器学习能扯上什么关系?都说“越熟悉越陌生”,这话放这一点儿不假;大部分人都接触或亲手绘制过热图,但在这个“过度包装”的时代(很多软件和在线工具都实现了“一...阅读全文>&g... 阅 读 全 部 >
原理提起热图,大家可能马上就能领会:热图不就是差异表达基因在不同分组样品中的表达水平吗?一般一列表示一个样品,一行表示一个基因,其中的颜色表示的相对表达值。但说到热图与机器学习,很多人都会惊讶:一个简单的热图与高大上的机器学习能扯上什么关系?都说“越熟悉越陌生”,这话放这一点儿不假;大部分人都接触或亲手绘制过热图,但在这个“过度包装”的时代(很多软件和在线工具都实现了“一...阅读全文>&g... 阅 读 全 部 >
原理提起热图,大家可能马上就能领会:热图不就是差异表达基因在不同分组样品中的表达水平吗?一般一列表示一个样品,一行表示一个基因,其中的颜色表示的相对表达值。但说到热图与机器学习,很多人都会惊讶:一个简单的热图与高大上的机器学习能扯上什么关系?都说“越熟悉越陌生”,这话放这一点儿不假;大部分人都接触或亲手绘制过热图,但在这个“过度包装”的时代(很多软件和在线工具都实现了“一...阅读全文>&g... 阅 读 全 部 >
原理提起热图,大家可能马上就能领会:热图不就是差异表达基因在不同分组样品中的表达水平吗?一般一列表示一个样品,一行表示一个基因,其中的颜色表示的相对表达值。但说到热图与机器学习,很多人都会惊讶:一个简单的热图与高大上的机器学习能扯上什么关系?都说“越熟悉越陌生”,这话放这一点儿不假;大部分人都接触或亲手绘制过热图,但在这个“过度包装”的时代(很多软件和在线工具都实现了“一...阅读全文>&g... 阅 读 全 部 >
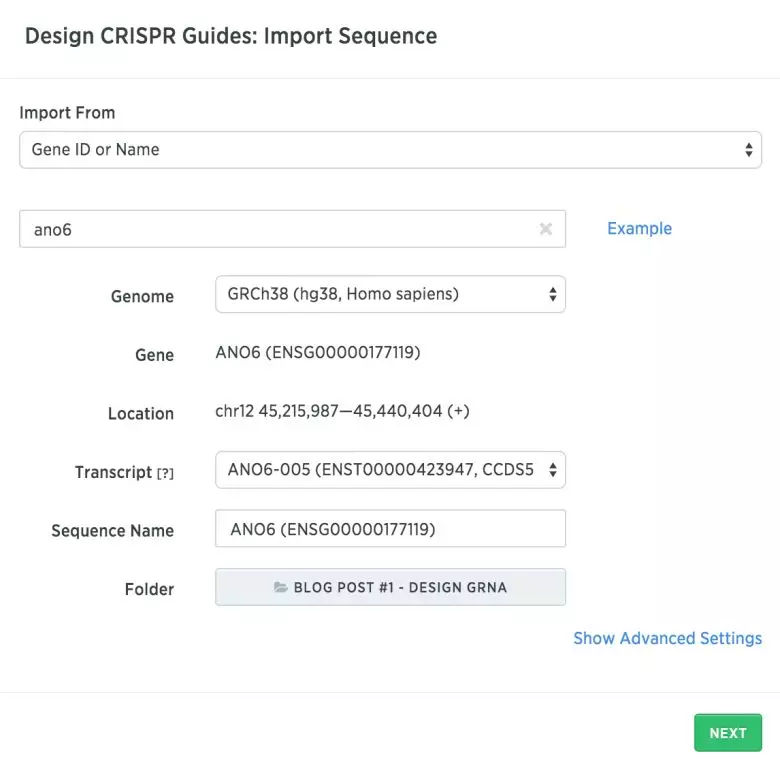 第一步 确定您想要定位的基因组区域您需要首先找到哪些外显子存在于您的靶基因的所有转录物中。要在Benchling中获取此信息,请点击右上角的“create”,然后点击“create CRISPR”。 在下一个对话框中,您可以搜索您感兴趣的基因。以下是在人类基因组(hg38)中搜索Ano6的示意图。在Advanced Settings中,您可以选择所有转录物以在基因组上应用注释。...阅读全文>... 阅 读 全 部 >
第一步 确定您想要定位的基因组区域您需要首先找到哪些外显子存在于您的靶基因的所有转录物中。要在Benchling中获取此信息,请点击右上角的“create”,然后点击“create CRISPR”。 在下一个对话框中,您可以搜索您感兴趣的基因。以下是在人类基因组(hg38)中搜索Ano6的示意图。在Advanced Settings中,您可以选择所有转录物以在基因组上应用注释。...阅读全文>... 阅 读 全 部 >
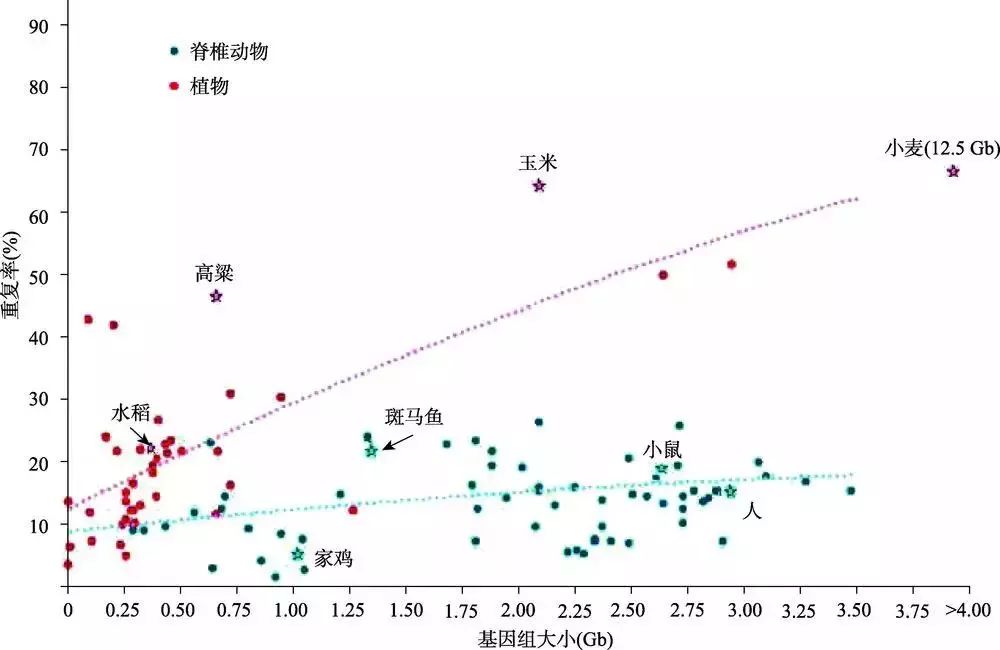 复杂基因组指的是无法使用常规测序和组装手段直接解析的一类基因组,通常指包含高比例重复序列、高杂合度、极端GC含量、存在难消除异源DNA污染的基因组。为了解决复杂基因组的测序和组装问题,需要分别从基因组测序实验方法、测序技术平台、组装算法与策略3个方面进行深入研究。本文详细介绍了复杂基因组测序组装相关的现有技术与方法,并结合复杂基因组经典实例介绍了复杂基因组测序的技术解决途径和发展历程,可为制订..... 阅 读 全 部 >
复杂基因组指的是无法使用常规测序和组装手段直接解析的一类基因组,通常指包含高比例重复序列、高杂合度、极端GC含量、存在难消除异源DNA污染的基因组。为了解决复杂基因组的测序和组装问题,需要分别从基因组测序实验方法、测序技术平台、组装算法与策略3个方面进行深入研究。本文详细介绍了复杂基因组测序组装相关的现有技术与方法,并结合复杂基因组经典实例介绍了复杂基因组测序的技术解决途径和发展历程,可为制订..... 阅 读 全 部 >
 利用生物信息分析大数据在论文发表中占据了举足轻重的地位,尤其是在高通量测序越来越便宜的今天,但是测序分析中各种名词仍令很多小菜或非生物信息专业的人抓狂。哈哈,不用怕,看了小编今天的文后,这些都不是事儿!先来介绍几个概念性名词:1.高通量测序: 高通量测序技术(High-throughput sequencing,HTS)是对传统Sanger测序(称为一代测序技术)革命性的改变, 一次对几十...阅... 阅 读 全 部 >
利用生物信息分析大数据在论文发表中占据了举足轻重的地位,尤其是在高通量测序越来越便宜的今天,但是测序分析中各种名词仍令很多小菜或非生物信息专业的人抓狂。哈哈,不用怕,看了小编今天的文后,这些都不是事儿!先来介绍几个概念性名词:1.高通量测序: 高通量测序技术(High-throughput sequencing,HTS)是对传统Sanger测序(称为一代测序技术)革命性的改变, 一次对几十...阅... 阅 读 全 部 >
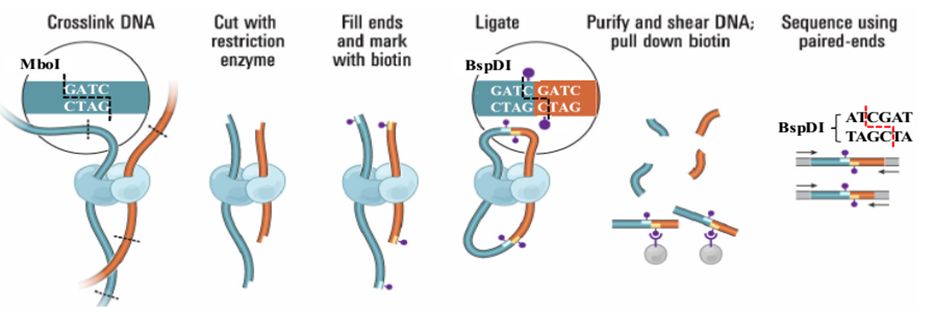 高质量的参考基因组是研究物种进化、性状定位、基因表达调控等生物学问题的基础,但目前二代+三代的测序策略,只能够将基因组组装到Contig/Scaffold水平,无法获得染色体水平的基因组信息。而 Hi-C辅助组装技术可将Contig/Scaffold挂载到不同的染色体上,提升基因组质量,在基因组文章的发表中扮演了不可或缺的角色。 下面,小编将与大家分享一些Hi-C辅助组装技术的小知识...阅读全文... 阅 读 全 部 >
高质量的参考基因组是研究物种进化、性状定位、基因表达调控等生物学问题的基础,但目前二代+三代的测序策略,只能够将基因组组装到Contig/Scaffold水平,无法获得染色体水平的基因组信息。而 Hi-C辅助组装技术可将Contig/Scaffold挂载到不同的染色体上,提升基因组质量,在基因组文章的发表中扮演了不可或缺的角色。 下面,小编将与大家分享一些Hi-C辅助组装技术的小知识...阅读全文... 阅 读 全 部 >
 植物基因组通常具有较高的重复序列,且很多为多倍体,因此组装植物基因组具有一定的挑战性。双子叶模式植物拟南芥、单子叶模式植物水稻基因组序列分别在2000年、2005年公布,它们都是基于BAC克隆及sanger法测序的方法获得的,至今在植物基因组序列中其质量依然是最好的。二代测序技术的出现及发展,极大地加快了植物基因组的研究进程,已经有超过200种植物获得了基因组序列,但是由于二代测序...阅读全文&... 阅 读 全 部 >
植物基因组通常具有较高的重复序列,且很多为多倍体,因此组装植物基因组具有一定的挑战性。双子叶模式植物拟南芥、单子叶模式植物水稻基因组序列分别在2000年、2005年公布,它们都是基于BAC克隆及sanger法测序的方法获得的,至今在植物基因组序列中其质量依然是最好的。二代测序技术的出现及发展,极大地加快了植物基因组的研究进程,已经有超过200种植物获得了基因组序列,但是由于二代测序...阅读全文&... 阅 读 全 部 >
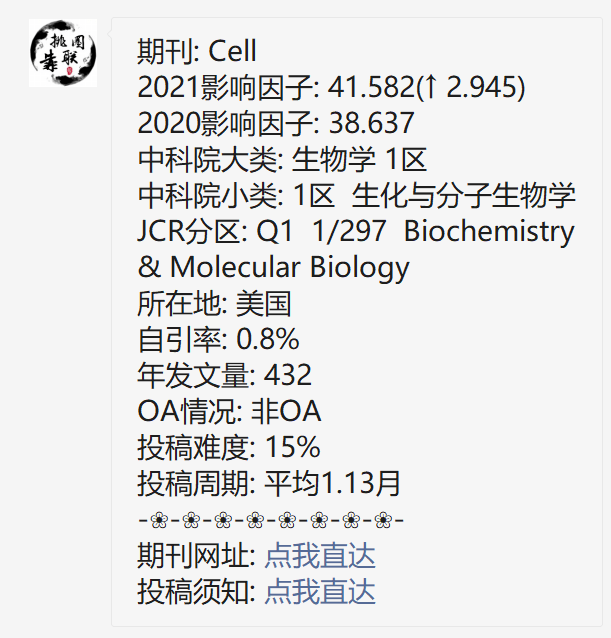 领略高端套路,发表高分文章!今天带给大家的是一篇于2021年1月发表在《Cell》上的高分生信文章,题目是“ A modular master regulator landscape controls cancer tranional identity”。 期刊简介数据来源 & 思路框架本项研究意在将基因组的改变与癌细胞转录的特性联系起... 阅 读 全 部 >
领略高端套路,发表高分文章!今天带给大家的是一篇于2021年1月发表在《Cell》上的高分生信文章,题目是“ A modular master regulator landscape controls cancer tranional identity”。 期刊简介数据来源 & 思路框架本项研究意在将基因组的改变与癌细胞转录的特性联系起... 阅 读 全 部 >
 领略高端套路,发表高分文章! 小伙伴们大家好,我是菠小萝。这里是菠小萝的高分生信SCI解读专栏。说到套路,大家都是又爱又恨,简单的套路分数低,分高的套路实验多,难道就没有我们自己也能通过基础的生信和简单的实验发表的高分文章吗?2020年的12月,菠小萝将以“你也能发表的高分生信”为专题,带给大家不一样的生信套路套路~本周我要与大家分享的是一篇2018年发表在《Cell Systems》上的纯生..... 阅 读 全 部 >
领略高端套路,发表高分文章! 小伙伴们大家好,我是菠小萝。这里是菠小萝的高分生信SCI解读专栏。说到套路,大家都是又爱又恨,简单的套路分数低,分高的套路实验多,难道就没有我们自己也能通过基础的生信和简单的实验发表的高分文章吗?2020年的12月,菠小萝将以“你也能发表的高分生信”为专题,带给大家不一样的生信套路套路~本周我要与大家分享的是一篇2018年发表在《Cell Systems》上的纯生..... 阅 读 全 部 >
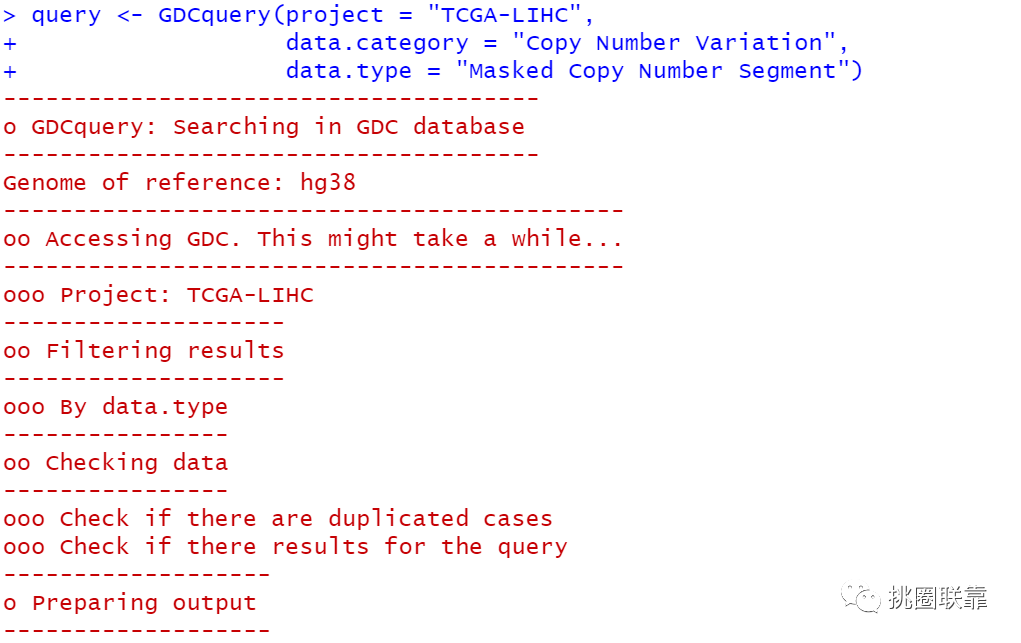 CNV变化的GISTIC_2.0分析大家好,我是阿琛。在基因组水平,除了单核苷酸多态性(single nucleotide polymorphism,SNP)水平的改变,拷贝数(Copy number variation, CNV)的变化同样是一个十分重要的改变因素。拷贝数变异,是由基因组发生重排而导致的, 一般指长度为 1 kb 以上的基因组大片段的拷贝数增加或者减少,主要表现为亚显微水平的缺失... 阅 读 全 部 >
CNV变化的GISTIC_2.0分析大家好,我是阿琛。在基因组水平,除了单核苷酸多态性(single nucleotide polymorphism,SNP)水平的改变,拷贝数(Copy number variation, CNV)的变化同样是一个十分重要的改变因素。拷贝数变异,是由基因组发生重排而导致的, 一般指长度为 1 kb 以上的基因组大片段的拷贝数增加或者减少,主要表现为亚显微水平的缺失... 阅 读 全 部 >

生信圈
欢迎您支持我的公众号
点击此处可关闭
 [该文章已设置加密,请点击标题输入密码访问]...
[该文章已设置加密,请点击标题输入密码访问]...