高质量的参考基因组是研究物种进化、性状定位、基因表达调控等生物学问题的基础,但目前二代+三代的测序策略,只能够将基因组组装到Contig/Scaffold水平,无法获得染色体水平的基因组信息。而 Hi-C辅助组装技术可将Contig/Scaffold挂载到不同的染色体上,提升基因组质量,在基因组文章的发表中扮演了不可或缺的角色。 下面,小编将与大家分享一些Hi-C辅助组装技术的小知识~
Hi-C辅助组装实验流程
利用甲醛对样本进行交联,质检合格后使用限制性内切酶(如MboI等)进行酶切,酶切片段经生物素标记、平末端连接、DNA纯化提取,超声打断后钓取含有生物素的片段,进行建库测序。随后,对原始下机数据进行质控,并将质控截取后的Clean reads与参考基因组比对,获得用于互作分析的Valid reads。由于Hi-C文库的构建具有一定的复杂性,在实际的项目执行过程中,会先通过对小规模的测序数据进行评估,以检测所构建文库的质量。小数据评估合格后,启动大数据的上机测序,以保证测序数据的质量。
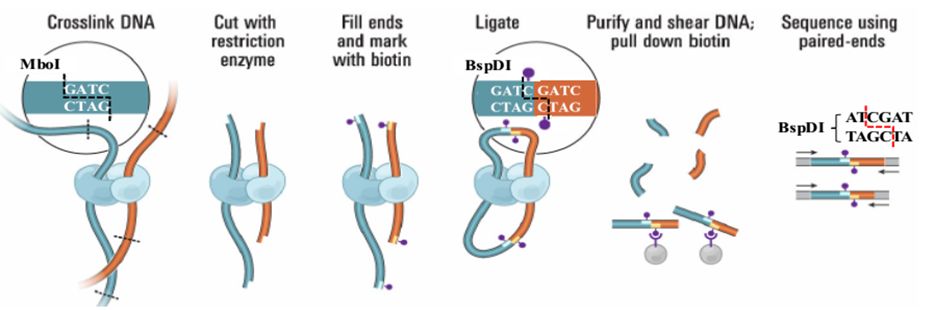
图1. Hi-C辅助组装实验流程图[1]
Hi-C数据处理
目前常用的Hi-C数据处理软件是HiC-Pro,该软件采用两步比对策略,有效提高了数据的利用率的同时,还提供了一系列的质控标准,对文库质量进行评估[2]。此外,HiC-Pro还具有从原始数据到归一化之后的Hi-C图谱构建的完整功能,内含迭代校正算法的优化版本,可显著加快和促进Hi-C数据的标准化。
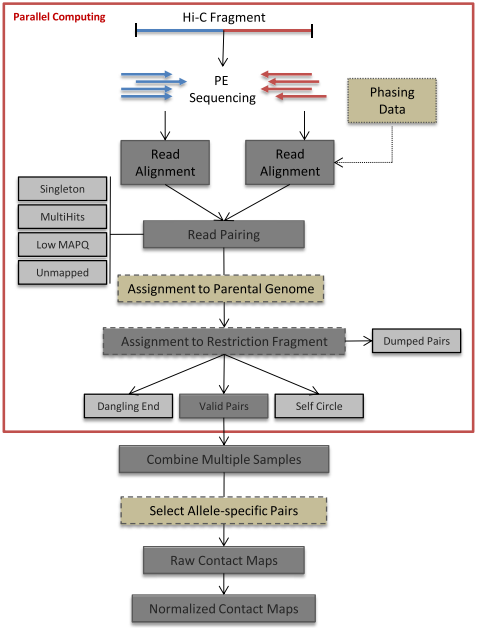
图2. HiC-Pro算法示意图[2]
Hi-C辅助组装常用软件
目前,用于Hi-C辅助基因组组装的软件有LACHESIS、SALSA2、3D-DNA、ALLHiC等,这些软件在基因组组装方面各有优劣。
LACHESIS
LACHESIS(ligating adjacent chromatin enables scaffolding in situ)是华盛顿大学于2013年研发的一种基因组组装方法,它通过聚类算法将初步组装的Contig/Scaffold分配到各染色体群中,基于染色体内部不同区段间的互作强度高低对每个染色体群组中的Contig/Scaffold进行排序和定向,将基因组草图提升到染色体水平[3]。LACHESIS是分析Hi-C数据的经典工具,目前已发表的Hi-C辅助组装文章多基于该算法,文章认可度较高。但LACHESIS在多倍体基因组组装方面具有一定的局限性,且该算法目前已停止更新。

图3. LACHESIS算法示意图[3]
SALSA2
SALSA是最早提出利用Hi-C对Contig进行纠错的软件,优化后的SALSA2提出了一种新的染色体划分方式,即检查每次迭代后是否存在错误的合并(mis-join),且该算法不需要预先设定染色体的数目,在一定程度上提高了Scaffold精确度[4]。但该算法会引入很多聚类/排序/定向错误,且在运行过程中可调参数较少,因此利用率一般。
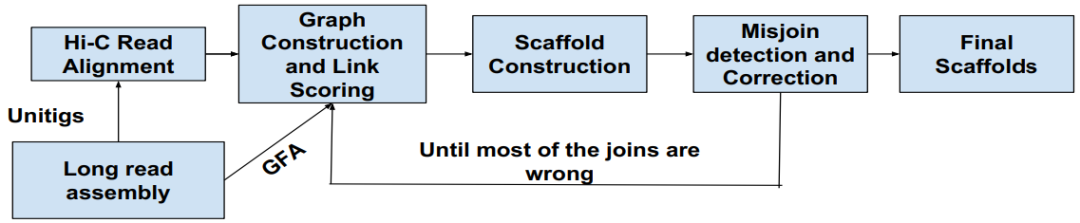
图4. SALSA2算法流程示意图[4]
3D-DNA
2017年Dudchenko等人通过3D-DNA(3D de novo assembly)实现了对基因组草图先纠错再组装的功能[5]。使用3D-DNA做基因组组装的整体流程包括组装、Juicer分析Hi-C数据、3D-DNA进行Scaffolding、使用JBAT对组装结果进行手工纠正,最终得到准染色体水平的基因组。但该软件的纠错功能并没有得到很好的应用:在模拟数据情况下3D-DNA的组装错误率是SALSA2错误率的2-4倍[4],且Juicer在多倍体物种上表现不佳。
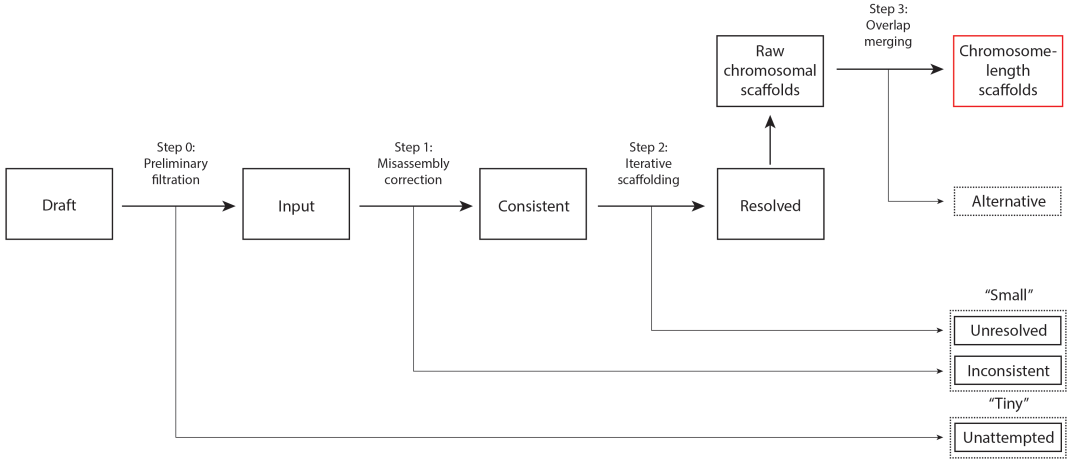
图5. 3D-DNA算法示意图[5]
ALLHiC
SALSA2、3D-DNA、LACHESIS等软件在动物基因组和简单植物基因组的Hi-C辅助组装上表现都不错,但在多倍体物种和高杂合物种中,由于等位基因序列的相似性,可能会使得不同染色体组之间的Contig出现假的互作信号,导致组装错误。而ALLHiC算法可通过修剪Hi-C平行信号和弱信号,将等位基因和同源序列分隔在不同的单倍型内独立组装,有效解决了多倍体物种和高杂合度基因组的辅助组装难题。此外,ALLHiC还通过优化算法,改进了Contig的排序和定向,尤其是连续性较低的Contig,极大地提高了短序列的排序和定向的准确性[6]。
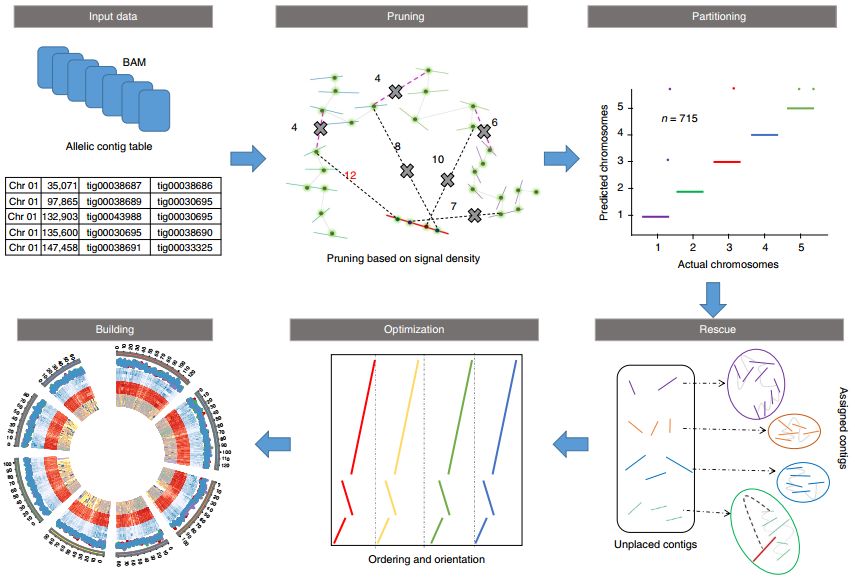
图6. ALLHiC算法示意图[6]
总结
SALSA2和3D-DNA虽不需预先提供染色体数目即可进行互作分析,但在可操作性和实用性上都有一定的局限性。LACHESIS作为分析Hi-C数据的经典工具,文章认可度较高,但其在多倍体基因组组装方面表现欠佳。相对而言,ALLHiC除了将等位基因和同源序列分隔在不同的单倍型内独立组装外,还通过算法优化,提高了对序列的排序和定向的准确性。因此,ALLHiC不但适用于多倍体基因组组装,还可用于简单的二倍体基因组、高杂合基因组组装。值得注意的是,LACHESIS和ALLHiC需预先提供准确的染色体数目,才能对Contig/Scaffold进行聚类、分群。
安诺基因在LACHESIS常用功能的基础上,通过对分析流程进行优化,增加了热图纠错功能,显著提升了实际项目中基因组的组装效果。在多倍体和高杂合基因组组装方面,安诺基因采用ALLHiC算法,为高质量基因组的获得提供保障。基于Hi-C辅助组装技术,安诺基因已与中国农业大学、中科院遗传与发育所、中国海洋大学、中国农业科学院、浙江大学、福建农林大学等多家科研院所开展了深度合作,助力基因组文章发表于Nature、Nature Plants、Nature Communications、Molecular Plant、Communications Biology、The Plant Journal 等多个国际高水平期刊。
参考文献
[1] Lieberman-Aiden E, Van Berkum N L, Williams L, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome[J]. Science, 2009, 326(5950): 289-293.
[2] Servant N, Varoquaux N, Lajoie B R, et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing[J]. Genome Biology, 2015, 16(1): 259.
[3] Burton J N, Adey A, Patwardhan R P, et al. Chromosome-scale scaffolding ofde novo genome assemblies based on chromatin interactions[J]. Nature Biotechnology, 2013, 31(12): 1119.
[4] Ghurye J, Rhie A, Walenz B P, et al. Integrating Hi-C links with assembly graphs for chromosome-scale assembly[J]. bioRxiv, 2019: 261149.
[5] Dudchenko O, Batra S S, Omer A D, et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds[J]. Science, 2017, 356(6333): 92-95.
[6] Zhang X, Zhang S, Zhao Q, et al. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data[J]. Nature Plants, 2019, 5(8): 833-845.- 本文固定链接: https://maimengkong.com/kyjc/1178.html
- 转载请注明: : 萌小白 2022年9月16日 于 卖萌控的博客 发表
- 百度已收录
