植物基因组通常具有较高的重复序列,且很多为多倍体,因此组装植物基因组具有一定的挑战性。双子叶模式植物拟南芥、单子叶模式植物水稻基因组序列分别在2000年、2005年公布,它们都是基于BAC克隆及sanger法测序的方法获得的,至今在植物基因组序列中其质量依然是最好的。
二代测序技术的出现及发展,极大地加快了植物基因组的研究进程,已经有超过200种植物获得了基因组序列,但是由于二代测序读长短,大部分的基因组组装结果都不高,含有数千个scaffolds;虽然基因区相对完整,但是富含转座元件的区域都装得比较碎,且明显低估了这些区域的比例,仅有少部分组装到了染色体水平。
近年来,随着三代PacBio、Oxford Nanopore测序技术的发展,可以获得较长的DNA片段,采用一定的组装软件,较为容易获得高质量的组装结果,尤其是在提高序列的完整性及重复序列组装方面有了很大的改善。不过到目前为止,依然只有少数植物基因组组装的完整性较好,contig N50>5Mb的只有6个;另外即使是基于长读长reads得到较长的contigN50(>1Mb),要想获得染色体水平的序列依然是不太容易的。

下面,小编通过Nature Plant一篇文献“Chromosome-scale assemblies of plant genomes using nanopore long reads and optical maps”了解下如何通过采用三代测序加上optical maps、Illumina二代数据及遗传图谱的策略获得染色体级别的植物基因组序列吧。
组装结果
文章研究了三个物种,分别为双子叶芸薹属的B. rapa(yellow sarson,Z1),B.oleracea(broccoli, HDEM)及单子叶芭蕉属的Musa schizocarpa(banana),这3个物种B.rapa Chiifu、B. oleracea To1000、Musa acuminate Pahang-HD曾采用short-reads策略获得了基因组序列,不过序列多为片段化(contig N50<50kb)。
文章中, B.rapa(Z1)、B.oleracea(broccoli, HDEM)、
M.schizocarpa分别用MinION测序获得了79X、32X、44X的数据,基于这些long-reads进行组装,得到了少于1000条contigs, contig N50在3.8到7.3Mb之间。再结合optical maps及Illumina short reads进行组装,最终contig N50在5.5到9.5Mb之间,scaffold N50在15.4到36.8Mb之间。1/4的染色体是单个scaffold,66%的染色体由1个或2个scaffolds组成,足见组装质量是相当好的。
本次组装contig N50与之前相比,提升了100倍和450倍;从注释结果来看,B. rapa,B. oleracea和M. schizocarpa分别注释出46,721,61,279,32,809个基因,与之前发表的结果相似。下表是本次组装结果与以往公布结果的详细比较。
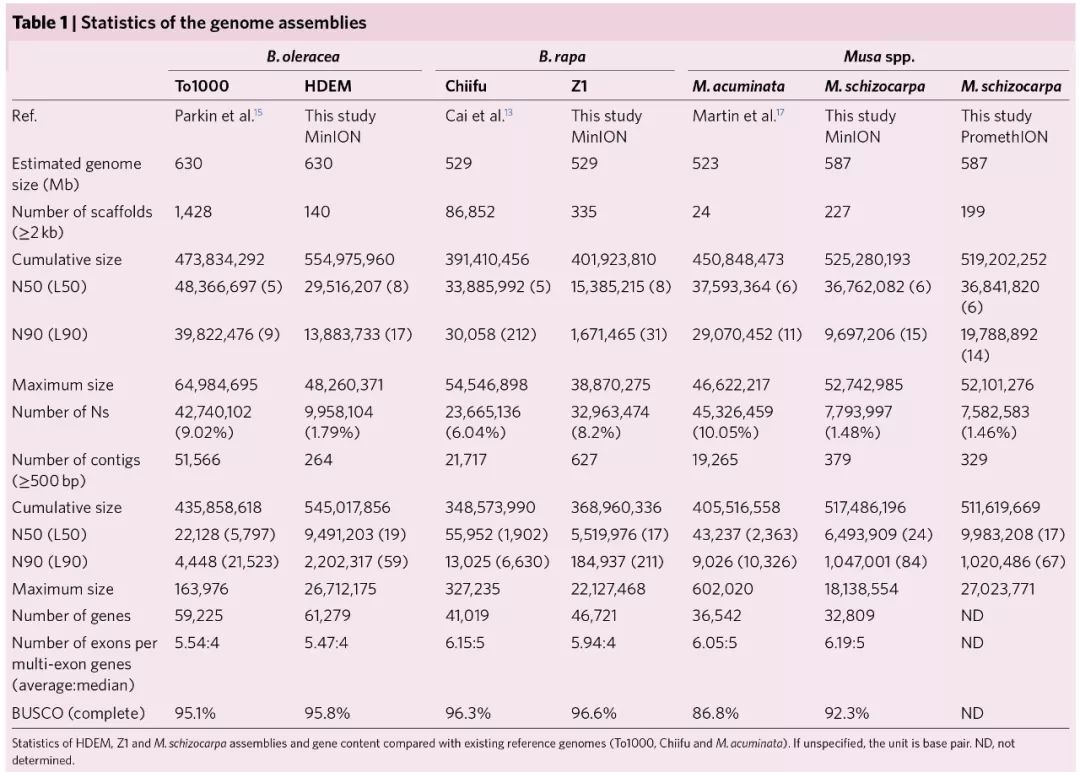
long read提升转座子富集区域的组装结果
值得一提的是,虽然注释的功能编码基因个数与之前发表的结果较一致,但是采用长片段测序明显提升了重复序列的组装结果,检测到的重复序列比例较高,且转座元件的平均长度要长;而且通常在转座子富集区域的基因难以定位到染色体上,但是本研究组装中能将超过98%的基因锚定到染色体上。由此可见,长reads对于提升转座元件富集区域的组装质量是非常关键的。
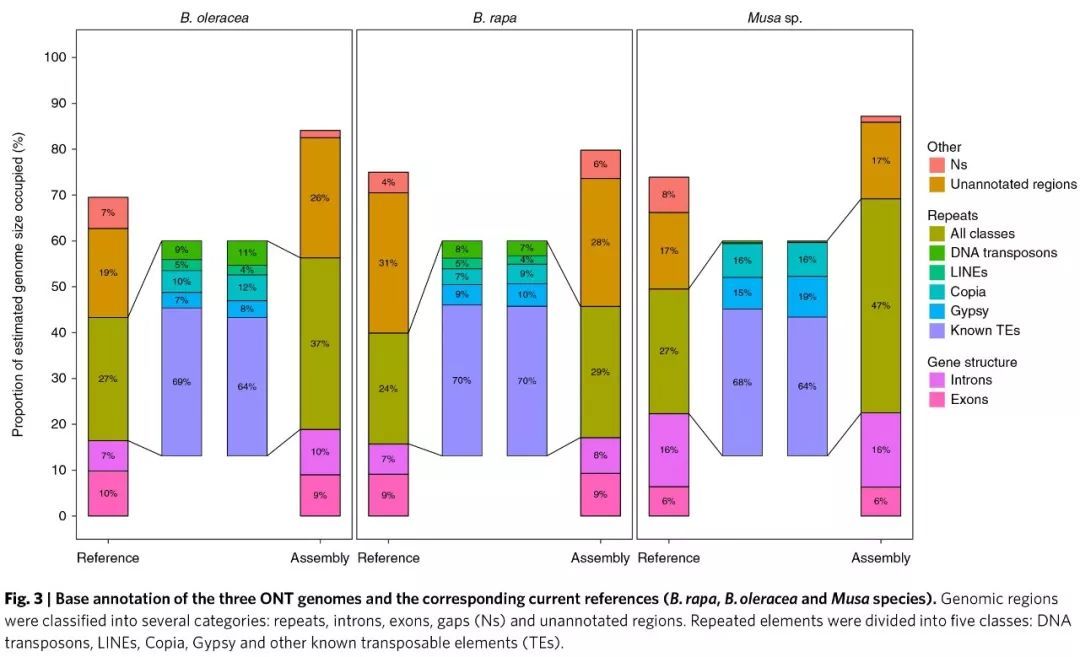
同时分析了FLC 基因的拷贝数(该基因与春化及开花时间有关,该基因家族的拷贝数变化能够影响开花时间),在B.oleracea(broccoli, HDEM) 和B.rapa Z1 中分别发现了7 个和4 个FLC 基因。表明长读长更有利于重复区域的组装。
抗病基因R-genes 一般是成簇出现的,较难正确地组装;the M. acuminata 和the M. schizocarpa 基因组中3 个同源R-gene 簇中不确定碱基的比例分别为6.5% 和0% ,再次显示了长读长对复杂区域组装的重要性。
进一步对199 份B.rapa 和119 份B.oleracea 材料进行了重测序,将测序结果与本次组装的序列及之前的序列进行比对,发现除了Chinese cabbage ,Chinese kale 外,其它的与本次组装的参考序列比对率更高;但是uniquely mapped reads 的比例要低,进一步说明新组装的序列重复区域组装的效果更好,新组装的序列更适合在后续芸薹属重测序中作为参考序列。

Nanopore数据、PacBio数据组装结果比较
PacBio测序、Nanopore测序都能获得长的reads,这两种测序技术对组装有何影响呢?文章比较了用PacBio测序的6个物种与本文的3个物种之间的组装结果(基因组大小在130-630Mb之间),发现使用ONT获得的大于50kb的reads比例更高;而PacBio的测序深度更高一些(在125X-283X之间),表明PacBio需要更高的测序深度以获得足够的长reads来提升组装的连续性。在这9个物种中,连续性第二好的,长读长reads的深度只有36X,但是reads长度是最长的,说明相比于测序深度,更长的reads对提升组装结果更有效;30X的long reads能够满足组装的需求。
不过,小编认为这里的比较如果在同一物种上进行平行比较会更有说服力,不过不管怎样,从文章的比较结果来看,reads长度是决定组装结果好坏的关键因素。

PacBio三代测序仪
随着Nanopore、PacBio测序技术的发展、升级及各种新组装软件的出现,必定会在极大降低研究费用的同时大大提升基因组组装效果,那些以往难以取得较好结果的复杂基因组,将会迎来研究契机,当然,那些相对简单的基因组组装结果也会更好。
微分基因通过搭建世界领先的高通量测序平台、基因芯片平台、大数据分析平台,在全国范围内与众多学术机构、研发企业以及健康管理平台建立了深入的合作关系,为一系列基因组科学研究、健康管理项目等提供专业基因测序技术服务。
如果您有物种想进行基因组测序,现在可以准备起来啦。
参考文献:
1.Arabidopsis Genome Initiative Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408, 796–815 (2000).
2.International Rice Genome Sequencing Project The map-based sequence of the rice genome. Nature 436, 793–800 (2005).
3.Du, H. et al. Sequencing and de novo assembly of a near complete indica rice genome. Nat. Commun. 8, 15324 (2017).
4.Edger, P. P. et al. Single-molecule sequencing and optical mapping yields an improved genome of woodland strawberry (Fragaria vesca) with chromosome-scale contiguity. Gigascience 7, 1–7 (2018).
5.Dassanayake, M. et al. The genome of the extremophile crucifer The llungiella parvula. Nat. Genet. 43, 913–918 (2011).
6.International Brachypodium Initiative Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature 463,763-768(2010)
7.Raymond, O. et al. The Rosa genome provides new insights into the domestication of modern roses. Nat. Genet. 50, 772–777 (2018).
8.Caroline B. et al. Chromosome-scale assemblies of plant genomes using nanopore long reads and optical maps. Nature Plant. 4(11):879–887 (2018)- 本文固定链接: https://maimengkong.com/kyjc/1177.html
- 转载请注明: : 萌小白 2022年9月10日 于 卖萌控的博客 发表
- 百度已收录
