CNV变化的GISTIC_2.0分析
大家好,我是阿琛。在基因组水平,除了单核苷酸多态性(single nucleotide polymorphism,SNP)水平的改变,拷贝数(Copy number variation, CNV)的变化同样是一个十分重要的改变因素。拷贝数变异,是由基因组发生重排而导致的, 一般指长度为 1 kb 以上的基因组大片段的拷贝数增加或者减少,主要表现为亚显微水平的缺失和重复。
GISTIC2分析,主要是用于检测一组样品中显着扩增或缺失的基因组区域,即通过分析每个样本的CNV检测结果,计算这一批样本中显著扩增和缺失的区域信息。一般而言,这个分析在癌症基因组CNV分析中十分常见也十分必要的内容。
下面,我们一起来看下如何进行基于CNV变化的GISTIC_2.0分析,包括输入文件准备和结果的可视化展示。
1. segment file数据下载和处理
1.1 从TCGA下载数据
这里,使用TCGAbiolinks包进行TCGA数据库中CNV数据的下载。
首先,通过GDCquery函数查询下载信息;
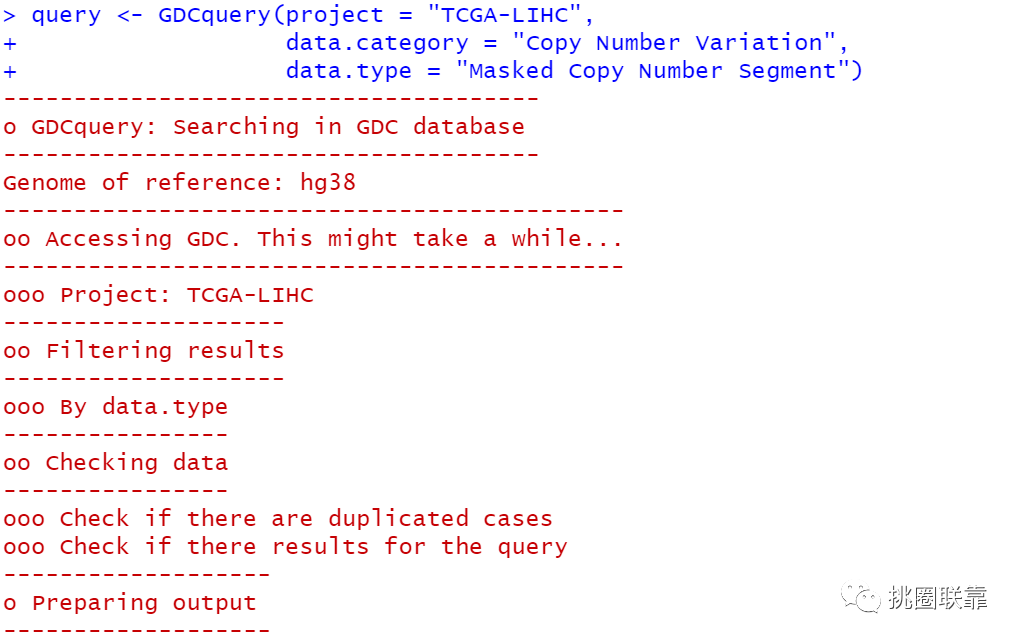
通过GDCdownload函数下载相应的CNV数据;
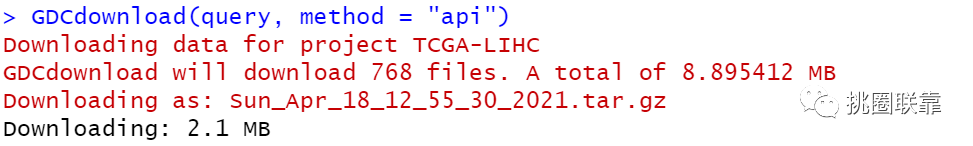
使用GDCprepare函数对下载得到的数据进行整合,并保存为.rda文件。
1.2 数据处理
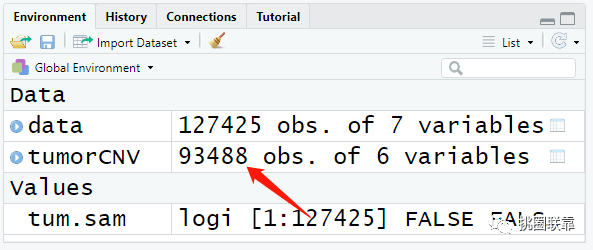
数据下载完成后,使用load函数读取保存的CNV数据,并赋值给变量tumorCNV。
随后,提取第2至7列的内容,包括样品名,染色体位置等信息;并对其顺序进行重新排列,将样品名字放到第一列。

1.3 提取肿瘤样本
接着,我们需要将其中的肿瘤样品进行提取。
结果显示:根据TCGA命名方式,其中包括肿瘤样品93488个结果。

最后,提取CNV数据中的肿瘤样品结果,并将其进行保存。这样,第一个输入文件就准备好了。
2. marker file数据下载和处理
2.1 从TCGA下载数据
接下来,我们需要准备第二份输入文件,即注释文件。
1. 进入T CGA数据库中注释文件的界面(https://gdc.cancer.gov/about-data/gdc-data-processing/gdc-reference-files);
2. 往下查找,可以看到SNP6 GRCh38 Remapped Probeset File for Copy Number Variation Analysis内容,其中包含了CNV数据的注释文件;点击“snp6.na35.remap.hg38.subset.txt.gz”进行下载;
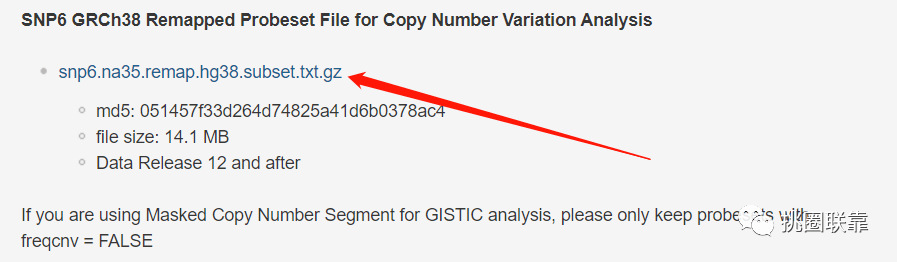
同时,我们可以看到下方的一段注意内容:
If you are using Masked Copy Number Segment for GISTIC analysis, please only keep probesets with freqcnv = FALSE
即:如果您使用CNV片段数据进行GISTIC分析,请仅使用freqcnv = FALSE保留探针集。因此,下载得到的注释文件需要进一步进行整理。
2.2 提取freqcnv=FALSE数据
首先,使用data.table包的fread函数读取注释文件。

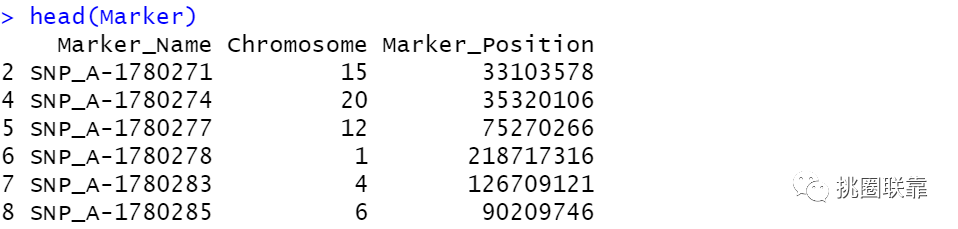
使用str函数查看一下注释文件Marer中包含的相关内容,结果显示:可以看到,其中包含了探针id,染色体的相关信息等内容。
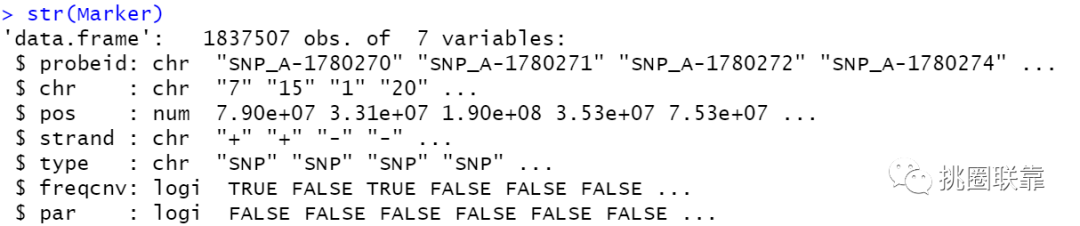
根据提示的内容,判断freqcnv列中为FALSE的内容。

最后,提取freqcnv为FALSE的行,对列名进行重新命名,并进行保存,作为第二个输入文件使用。
3. GenePattern GISTIC_2.0在线分析
接下来,我们进行GISTIC 2.0分析。关于GISTIC 2.0分析,存在在线分析和离线版两种。这里,我们介绍一下在线版GISTIC_2.0分析方法。
1. 首先,进入GenePattern网站(https://cloud.genepattern.org/gp/pages/index.jsf);对于首次进入GenePattern网站的话,使用邮箱进行注册即可;
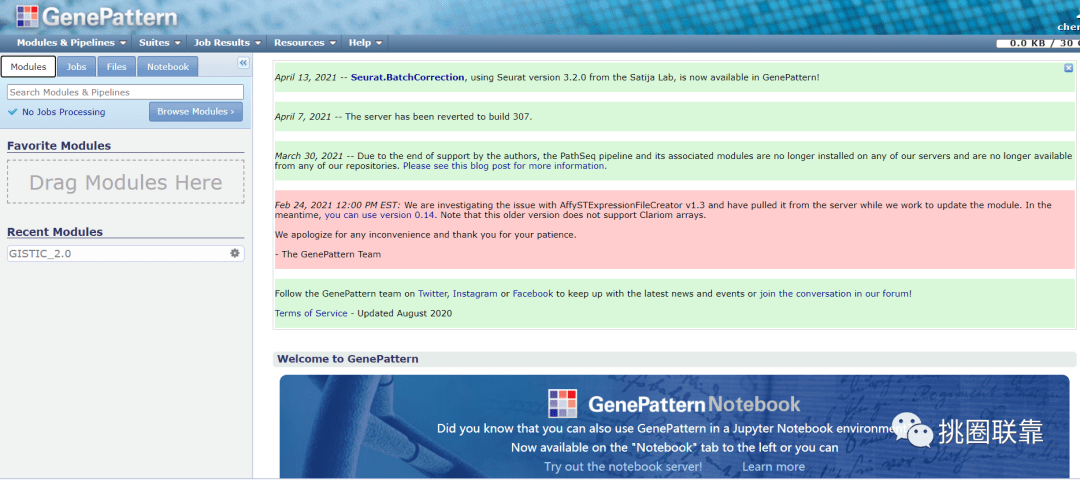
2. 在模块Modules下,点击Browse Modules,选择All Modules,进入模块选择板块;
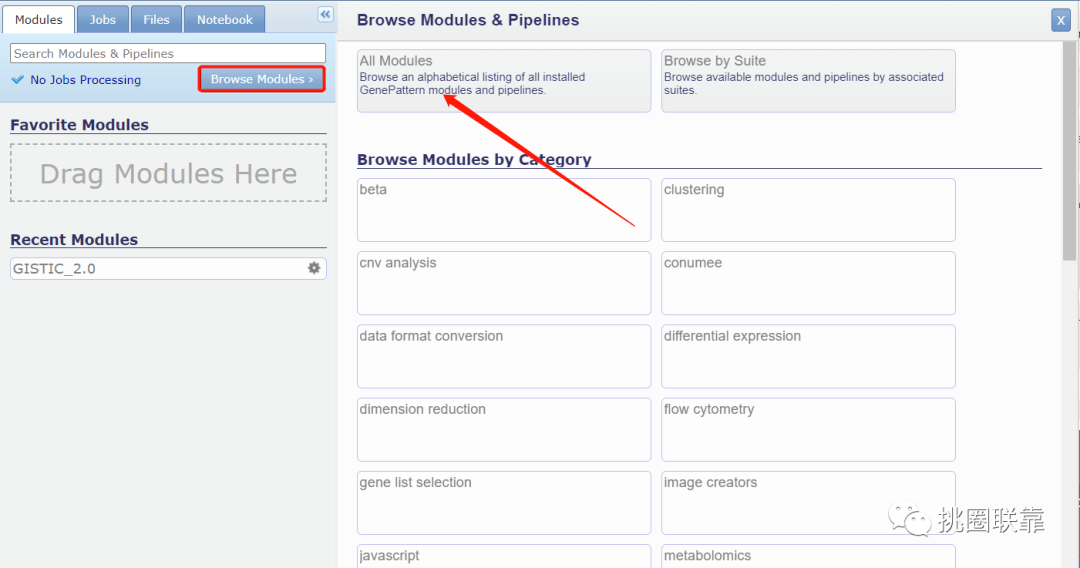
3. 查找GISTIC_2.0分析模块,点击选中;
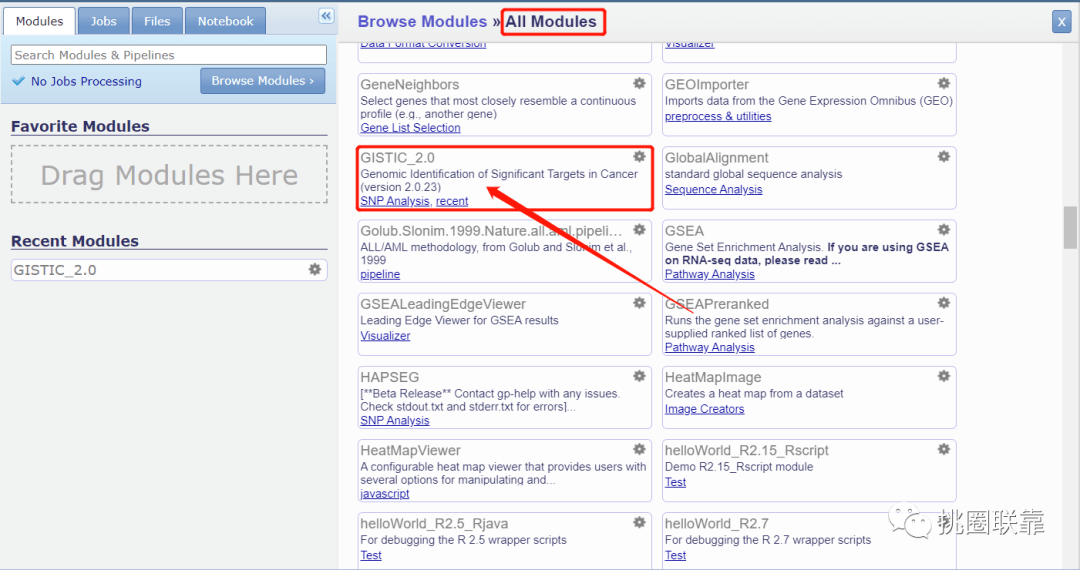
此时,我们就进入了GISTIC_2.0分析模块中;
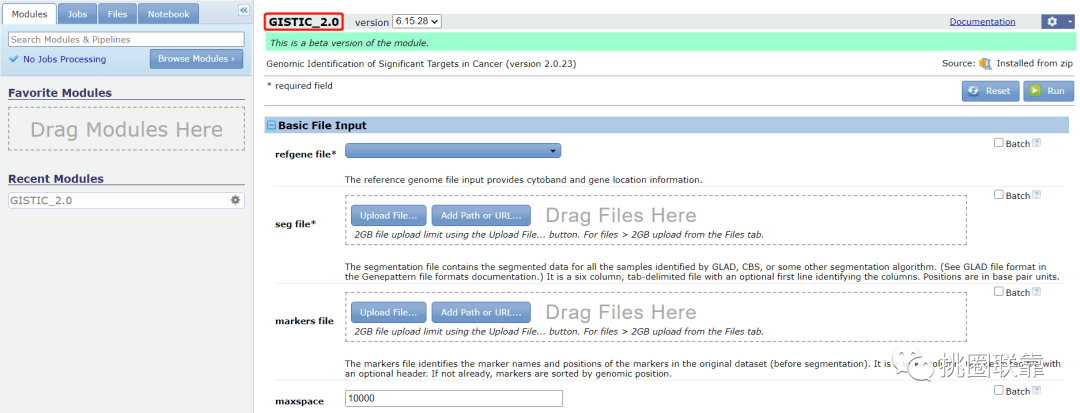
4.根据要求,对于TCGA数据分析,在refgene.file中选
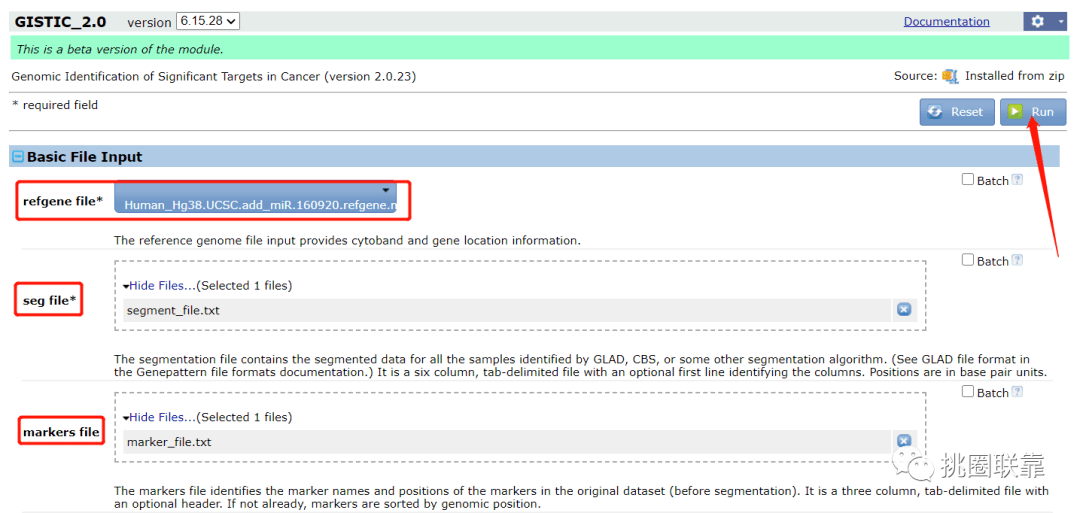
5.随后,进行运行阶段,大约需要耗时30min左右;勾选Email Reminder,在运行完成后可以收到相应的邮件;

6.待运行完成后,选择下载文件,即可获得相应的输出文件;
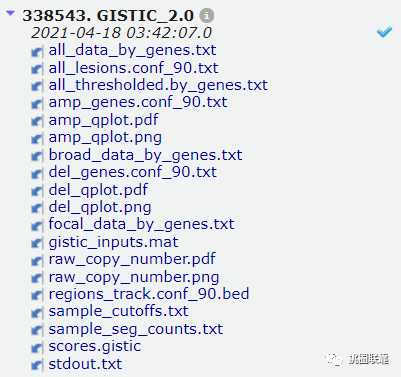
4. maftools可视化GISTIC结果
将GISTIC_2.0分析得到的结果文件保存到工作目录下,接下来,我们对结果进行可视化展示。
这里,我们使用maftools包进行可视化分析。
根据运行结果,依次将结果赋值给相应的参数,以构建GISTIC输入文件。
接下来,对结果进行可视化展示。
结果显示:其中红色表示拷贝数增加,而蓝色表示拷贝数降低,并对其中几个较为显著的基因名字信息进行了标注。
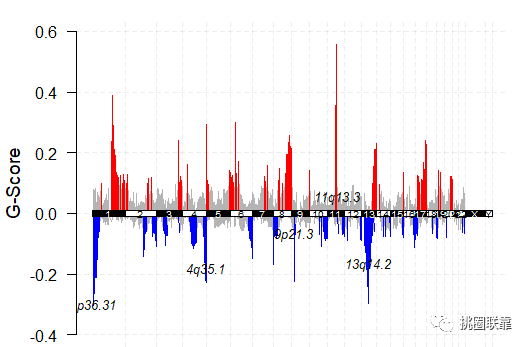
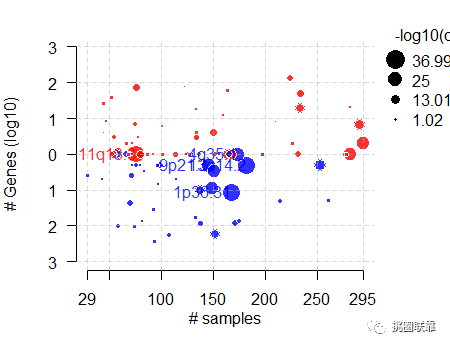
结果显示:
好啦,今天的分享就到此为止了~又get一项新的技能。赶紧根据示例代码和数据实践起来吧。
- 本文固定链接: https://maimengkong.com/kyjc/1173.html
- 转载请注明: : 萌小白 2022年9月3日 于 卖萌控的博客 发表
- 百度已收录
