2023
04-23
04-23
群体遗传分析—LD连锁不平衡
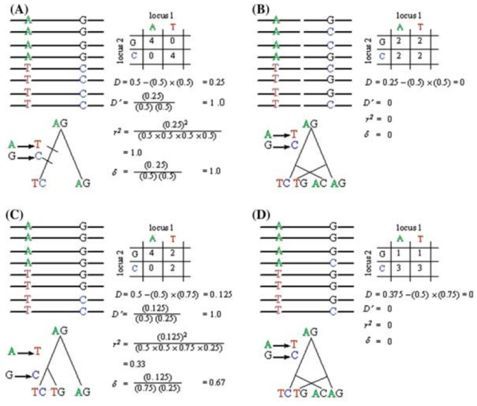 群体遗传学研究中,LD连锁不平衡分析是最常见的分析内容,也是关联分析的基础。在很多的遗传进化GWAS的文章中都会出现LD衰减及单体型block图,如果你还不是很了解的话,是时候补补课了哦~~LD概念当位于某一座位的特定等位基因与另一座位的某一等位基因同时出现的概率大于群体中因随机分布的两个等位基因同时出现的概率时,就称这两个座位处于连锁不平衡状态(linkage disequilibri...阅读... 阅 读 全 部 >
群体遗传学研究中,LD连锁不平衡分析是最常见的分析内容,也是关联分析的基础。在很多的遗传进化GWAS的文章中都会出现LD衰减及单体型block图,如果你还不是很了解的话,是时候补补课了哦~~LD概念当位于某一座位的特定等位基因与另一座位的某一等位基因同时出现的概率大于群体中因随机分布的两个等位基因同时出现的概率时,就称这两个座位处于连锁不平衡状态(linkage disequilibri...阅读... 阅 读 全 部 >
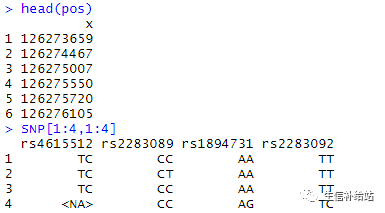 连锁不平衡图,用来可视化不同SNP之间的连锁程度,前同事间俗称“倒三角”图。本文使用自己的数据,因为安装R包后使用内置数据集运行出结果较容易,但是自己的数据就可能会有一些不大不小的“坑”,我替你们趟了。。。一 、载入R包数据数据为内置CEUData保存后,进行了“细微”的处理(去掉SNP碱基之间的“/”),因为这种基因型形式文件很常见;library("LDheatmap")#读入数据SNP .....
连锁不平衡图,用来可视化不同SNP之间的连锁程度,前同事间俗称“倒三角”图。本文使用自己的数据,因为安装R包后使用内置数据集运行出结果较容易,但是自己的数据就可能会有一些不大不小的“坑”,我替你们趟了。。。一 、载入R包数据数据为内置CEUData保存后,进行了“细微”的处理(去掉SNP碱基之间的“/”),因为这种基因型形式文件很常见;library("LDheatmap")#读入数据SNP .....  由于文章的需要,需要对LD block进行修改,之前是用HaploView进行绘制的,但是调整不了出图参数,故尝试使用R包LDheatmap重新绘制。首先是LDheatmap的安装,由于其依赖包 snpStats在Bioconductor 上,故先安装该包,再安装 LDheatmapif (!requireNamespace("BiocManager",...阅读全文>>...
由于文章的需要,需要对LD block进行修改,之前是用HaploView进行绘制的,但是调整不了出图参数,故尝试使用R包LDheatmap重新绘制。首先是LDheatmap的安装,由于其依赖包 snpStats在Bioconductor 上,故先安装该包,再安装 LDheatmapif (!requireNamespace("BiocManager",...阅读全文>>... 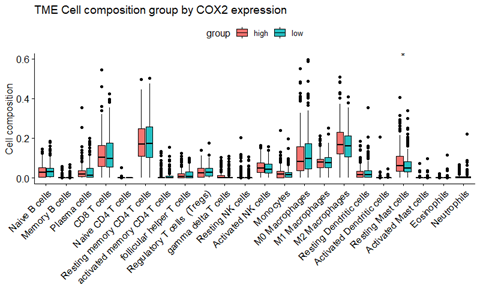 总结目前主流的免疫细胞浸润方法,整理归纳可用的资源及相关代码的实现,以及可视化方式,方便以后查询和分享。这里面提到的免疫浸润分析主要是基于组织样本转录组测序数据或者基因芯片数据的分析。因为我们取到的肿瘤组织并不只是包含肿瘤细胞,其中还有正常细胞、免疫细胞、基质细胞、血管细胞等。不同的细胞具有一些标志性的marker,免疫细胞也是一样。因此,可以根据这些marker基因在组织中的表达...阅读全文&...
总结目前主流的免疫细胞浸润方法,整理归纳可用的资源及相关代码的实现,以及可视化方式,方便以后查询和分享。这里面提到的免疫浸润分析主要是基于组织样本转录组测序数据或者基因芯片数据的分析。因为我们取到的肿瘤组织并不只是包含肿瘤细胞,其中还有正常细胞、免疫细胞、基质细胞、血管细胞等。不同的细胞具有一些标志性的marker,免疫细胞也是一样。因此,可以根据这些marker基因在组织中的表达...阅读全文&... 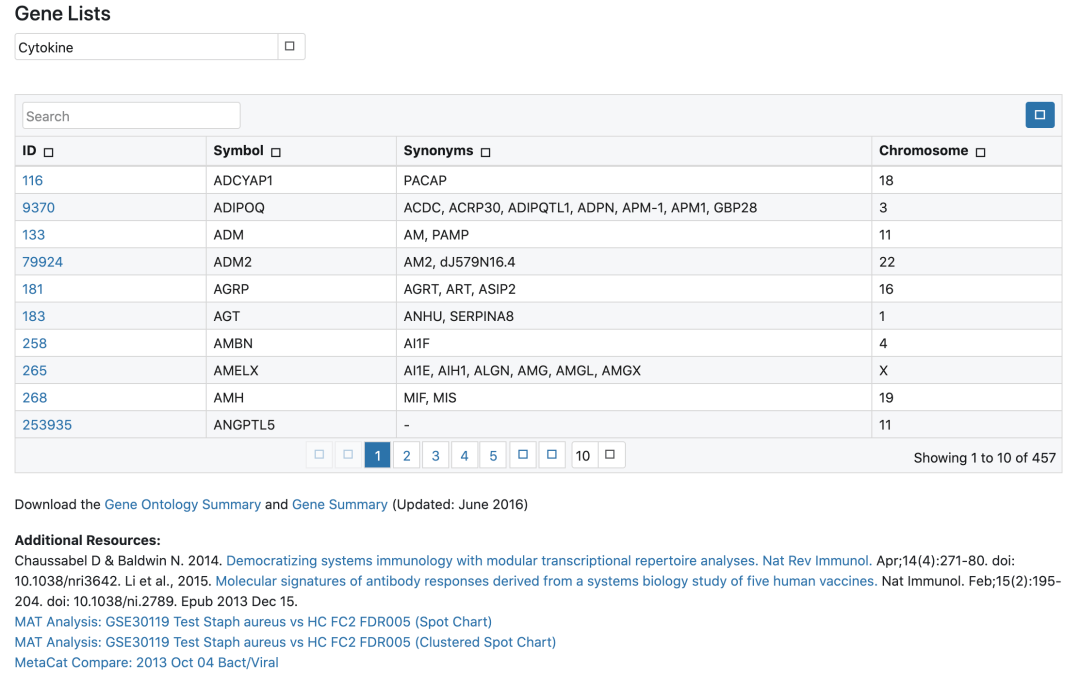 种数据挖掘文章本质上都是要把目标基因集缩小,比如表达量矩阵通常是2万多个蛋白编码基因,不管是表达芯片还是RNA-seq测序的,采用何种程度的差异分析,最后都还有成百上千个目标基因。如果是临床队列,通常是会跟生存分析进行交集,或者多个数据集差异结果的交集,比如:多个数据集整合神器-RobustRankAggreg包 ,这样的基因集就是100个以内的数量了,但是仍然有缩小的空间,比如lasso等统计学...
种数据挖掘文章本质上都是要把目标基因集缩小,比如表达量矩阵通常是2万多个蛋白编码基因,不管是表达芯片还是RNA-seq测序的,采用何种程度的差异分析,最后都还有成百上千个目标基因。如果是临床队列,通常是会跟生存分析进行交集,或者多个数据集差异结果的交集,比如:多个数据集整合神器-RobustRankAggreg包 ,这样的基因集就是100个以内的数量了,但是仍然有缩小的空间,比如lasso等统计学...  2019年07月05日,Nature Communications在线发表了中国农业科学院棉花研究所与北京百迈客生物科技有限公司共同合作文章“Extensive intraspecific gene order and gene structural variations in uplandcotton cultivars”详细研究内容如下:英文题目:Extensive intraspecific...
2019年07月05日,Nature Communications在线发表了中国农业科学院棉花研究所与北京百迈客生物科技有限公司共同合作文章“Extensive intraspecific gene order and gene structural variations in uplandcotton cultivars”详细研究内容如下:英文题目:Extensive intraspecific...  karyoploteR是一个bioconductor的一个R包,用于定制非环形全基因组数据的可视化。其绘图绘图过程按照R的基本绘图系统,并且不需要其他图形包。karyoploteR旨在给使用者提供一种可以创建任何线性染色体基因组的表征,并在染色体上绘制相关基因组注释和实验数据。 karyoploteR仅仅只是一个绘图工具,因此意味着无法用这个R包来下载数据,需要使用者自己去下载然后导入R...阅...
karyoploteR是一个bioconductor的一个R包,用于定制非环形全基因组数据的可视化。其绘图绘图过程按照R的基本绘图系统,并且不需要其他图形包。karyoploteR旨在给使用者提供一种可以创建任何线性染色体基因组的表征,并在染色体上绘制相关基因组注释和实验数据。 karyoploteR仅仅只是一个绘图工具,因此意味着无法用这个R包来下载数据,需要使用者自己去下载然后导入R...阅...  tructure是由斯坦福大学Pritchard实验室开发的一款群体结构分析软件。structure可通过贝叶斯聚类方法对每个样本的来源进行判断,进而反应群体的遗传结构。真正跑时burnin设为100000,跑100000代,k设为1-20,每轮运行10次。序列文件用xmfa2struct转为structure可识别的格式,SSR用Genalex转为structure可识别的格式。操作步骤打开st...
tructure是由斯坦福大学Pritchard实验室开发的一款群体结构分析软件。structure可通过贝叶斯聚类方法对每个样本的来源进行判断,进而反应群体的遗传结构。真正跑时burnin设为100000,跑100000代,k设为1-20,每轮运行10次。序列文件用xmfa2struct转为structure可识别的格式,SSR用Genalex转为structure可识别的格式。操作步骤打开st...  系统发育树系统发育树(phylogenetic tree),又称系统发生树、演化树、进化树、系统树、系统演化树、种系发生树等。表明被认为具有共同祖先的各物种之间的演化关系树形图,用来描述物种之间的分类和演化关系。自达尔文时期,很多生物学家希望用一棵树的形式,描述地球上各个物种的演化史。最理想的方法,就是采集各个物种的化石(直接证据),基于化石信息构建系统树。但是最大的缺点就是化石零散、不完整,甚至...
系统发育树系统发育树(phylogenetic tree),又称系统发生树、演化树、进化树、系统树、系统演化树、种系发生树等。表明被认为具有共同祖先的各物种之间的演化关系树形图,用来描述物种之间的分类和演化关系。自达尔文时期,很多生物学家希望用一棵树的形式,描述地球上各个物种的演化史。最理想的方法,就是采集各个物种的化石(直接证据),基于化石信息构建系统树。但是最大的缺点就是化石零散、不完整,甚至...  一直关注联川公众号的小伙伴们都知道,联川云平台已于2018年12月6日正式上线( http://cloud.lc-bio.com:5390/index.html);还没使用过的小伙伴,赶紧点开大显身手一番~使用指南详情请戳此链接:联川生物云平台使用指南;联川云平台包含FAQ/SOP和云平台双重功能:丰富的FAQ/SOP,助你快速上手入门,更有详尽的分析技能小技巧等你来学习;云平台中包含科研中经常用...
一直关注联川公众号的小伙伴们都知道,联川云平台已于2018年12月6日正式上线( http://cloud.lc-bio.com:5390/index.html);还没使用过的小伙伴,赶紧点开大显身手一番~使用指南详情请戳此链接:联川生物云平台使用指南;联川云平台包含FAQ/SOP和云平台双重功能:丰富的FAQ/SOP,助你快速上手入门,更有详尽的分析技能小技巧等你来学习;云平台中包含科研中经常用... 