大多数生物信息初学者最早接触的生物信息分析流程,大多数是用脚本语言(Perl、shell、R、Python等)将生物信息流程中的各个功能写成一个个脚本,再用大脚本套小脚本的方式将整个流程打包封装。这种方式对编程能力有很高的要求,否则很容易出现流程bug、无法监控任务运行状态、无法有效查看任务日志等问题。等实验室终于出了个大牛将这些问题都解决了,可能大牛又要毕业了,新来的生信小白又只能看着层层叠叠的嵌套脚本“望洋兴叹”……
这时候就需要 Snakemake 闪亮登场了,因为snakemake可能是处理这类问题最简单、有效的一个方法了。比如将流程像下图这样搭建起来,清晰明了:
PS:这张流程图也是 snakemake 通过命令自动生成的奥,具体命令后面会有介绍。
Snakemake是一个工作流程管理系统。它是基于Python的、用于创建可重现和可扩展的数据分析的工具(当然现在也可以直接将它当做Python的一个模块)。Snakemake所创建的流程还可以无缝扩展到服务器、集群和云环境等不同环境,当然前提是你需要提前将所需的软件和依赖配置好,一起打包封装在conda环境中。
snakemake的用法介绍
Snakemake 是基于 Python 的一款工具,所以它也继承了 Python 语言简单易读、逻辑清晰、便于维护的特点,同时它还支持 Python 语法,非常适合新手用户。snakemake 的基本组成单位叫“规则”,即 rule;每个 rule 里面又有多个元素(input、output、run等)。它的执行逻辑就是将各个 rule 利用 input/output 连接起来,形成一个完整的工作流,当检测到 input,就执行相应 rule;检测到 output,就跳过相应rule,根据这一规则,snakemake 还可以实现断点续投。
进阶拓展:如果你想要更强大的功能,可以在 rule 里面添加更多的元素,例如:
threads:设置核心数,来指定程序运行的线程
resources:指定程序运行的内存
log:指定生成日志文件
conda:指定专用的 conda 环境
更多规则元素,请参考官方文档https://snakemake.readthedocs.io/en/stable/snakefiles/rules.html
话不多说,让我们以一个变异检测流程为例,开始实操吧。
01
操作环境
本教程全程在Linux环境下完成,为了顺利完成教程内容,请准备一个Linux环境。如果是windows用户,可以安装一个虚拟机;如果是win10用户,可以使用win10自带的子系统(WSL)安装Linux系统。
02
软件安装
这里建议使用conda安装软件,可以轻松解决各种依赖问题。同时建议将本教程中用到的软件安装在一个新的conda环境中,以避免不同版本软件间的依赖冲突。由于篇幅有限,conda的安装方法不再赘述。
condacreate -n snakemake_env python snakemake
该命令创建了一个名为snakemake_env的conda环境,并在该环境中安装了 snakemake 。
#进入 snakemake_env 环境
conda activate snakemake_env
#退出 snakemake_env 环境
conda deactivate
进入 snakemake_env 环境,查看上面要安装的软件,可以发现都已将安装完成。
$snakemake -v
5.3.0
$python --version
Python 3.6.10 :: Anaconda, Inc.
如果你的conda安装很慢或者获取不了最新版本的软件,可以使用下面的命令安装:
$conda install -c conda-forge mamba
$mamba create -c conda-forge -c bioconda -n snakemake_env python snakemake
$conda activate snakemake_env
$snakemake -- help
03
数据准备
我们直接下载官网提供的测试数据:
$mkdir snakemake-demo
$cdsnakemake-demo
$wget https://github.com/snakemake/snakemake-tutorial-data/archive/v5.4.5.tar.gz
$tar --wildcards -xf v5.4.5.tar.gz --strip 1 "*/data"
解压后得到一个 data 文件夹,目录如下:
data
├── genome.fa
├── genome.fa.amb
├── genome.fa.ann
├── genome.fa.bwt
├── genome.fa.fai
├── genome.fa.pac
├── genome.fa.sa
└── samples
├── A.fastq
├── B.fastq
└── C.fastq
04
创建工作流文件:Snakefile
#创建 Snakefile
$touch Snakefile
# dry run(不真正执行,只会显示执行内容和顺序)
#目前 Snakefile 是空文件,所以显示如下所示
$snakemake -n
Building DAG of jobs...
Nothing to be done.
当然工作流文件 Snakefile 文件名并不是固定的,位置也不是固定的。当你的工作流文件就在当前目录下,且名称正好是 Snakefile 时,就可以像上面的示例一样,不用指定具体的工作流文件,snakemake 会自动调取当前路径的名为 Snakefile 的文件去执行。如果你要执行其他路径的 Snakefile 或者其他的文件名的工作流文件,可以使用 -s 参数,例如:
PS:建议将工作流文件命名为 py 文件,这样你在写 Python 代码时会有语法高亮奥。
$snakemake -n -s test.py
Building DAG of jobs...
Nothing to be done.
05
创建第一条规则 bwa
rule bwa: # 定义第一条规则,命名为 bwa
input: # input: 指定输入
"data/genome.fa",
"data/samples/A.fastq"
output: # output: 指定输出
"mapped/A.bam"
shell: # 指定执行方式,这里有三种执行方式:shell、run、,分别对应shell命令、Python命令、已经写好的脚本
"bwa mem {input} | samtools view -Sb - > {output}"
我们继续 dry run 一下,可以加上 -p 参数让终端打印出 shell 运行的命令:
$snakemake -np -s test.py
Building DAG of jobs...
Job counts:
count jobs
1 bwa
1
[Tue Aug 25 16:35:25 2020]
rule bwa:
input: data/genome.fa, data/samples/A.fastq
output: mapped/A.bam
jobid: 0
bwa mem data/genome.fa data/samples/A.fastq | samtools view -Sb - > mapped/A.bam
Job counts:
count jobs
1 bwa
1
06
使用通配符升级规则
上面的 bwa 规则仅仅比对了一个样本,可是实际项目中有几十上百的样本时,我们就不能这样直接写样本名来运行了,这样很不 pythonic。snakemake 允许使用通配符(wildcard)来批量运行命令,示例如下:
rule bwa:
input:
"data/genome.fa",
"data/samples/{sample}.fastq"
output:
"mapped/{sample}.bam"
shell:
"bwa mem {input} | samtools view -Sb - > {output}"
其中,我们使用通配符 {sample} 来匹配所有样品,花括号里的 sample 也可以使用其他字符。然后再次 dry run:
#这里需要加上规则的输出才能让含有通配符的程序正常运行
$snakemake -np -s test.py mapped/{A,B,C}.bam
Building DAG of jobs...
Job counts:
count jobs
3 bwa
3
[Tue Aug 25 16:40:32 2020]
rule bwa:
input: data/genome.fa, data/samples/A.fastq
output: mapped/A.bam
jobid: 0
wildcards: sample=A
bwa mem data/genome.fa data/samples/A.fastq | samtools view -Sb - > mapped/A.bam
[Tue Aug 25 16:40:32 2020]
rule bwa:
input: data/genome.fa, data/samples/B.fastq
output: mapped/B.bam
jobid: 1
wildcards: sample=B
bwa mem data/genome.fa data/samples/B.fastq | samtools view -Sb - > mapped/B.bam
[Tue Aug 25 16:40:32 2020]
rule bwa:
input: data/genome.fa, data/samples/C.fastq
output: mapped/C.bam
jobid: 2
wildcards: sample=C
bwa mem data/genome.fa data/samples/C.fastq | samtools view -Sb - > mapped/C.bam
Job counts:
count jobs
3 bwa
3
07
添加 sort 规则
比对完成之后需要使用 samtools 进行排序,那我们就可以继续写下一个规则:
rule sort:
input:
"mapped/{sample}.bam"
output:
"mapped/{sample}.sorted.bam"
shell:
"samtools sort -o {output} {input}"
08
建立索引
rule samtools_index:
input:
"mapped/{sample}.bam"
output:
"mapped/{sample}.bam.bai"
shell:
"samtools index {input}"
09
添加 calling 规则
前面三步都是各个样品批量、并行运行同样的步骤,所以可以全部使用通配符 {sample} 完成匹配;但在变异检测这一步需要将所有样本的bam文件传递给一个命令而不再并行,这种方法就不在适用了。针对这种情况,snakemake 有它独特的解决办法:
1. 首先在工作流文件开头定义一个变量:
samples = [ "A", "B", "C"]
2. 然后使用 snakemake 的内置函数 expand:
bam=expand( "mapped/{sample}.sorted.bam", sample=samples)
PS:这个 expand 函数其实是 Python 的列表推导式转化而来,有兴趣的同学可以自己研究一下。
最后,calling 规则如下:
rule calling:
input:
fa= "data/genome.fa",
bam=expand( "mapped/{sample}.sorted.bam", sample=samples),
bai=expand( "mapped/{sample}.sorted.bam.bai", sample=samples)
output:
"calling/all.vcf"
shell:
"samtools mpileup -g -f {input.fa} {input.bam} | "
"bcftools call -mv - > {output}"
10
调用自己写的脚本
当然,有时候在某个步骤想实现的功能并不能用简单的几句命令来实现,而且你自己也已经有了一个现成的脚本了,那么你就可以直接调用该脚本实现相应功能:
rule stats:
input:
"calling/all.vcf"
output:
"stat/stat.txt"
:
"s/stat.py"
11
最重要的一条规则:all
前面我们已经讲过,snakemake 通过各个规则的输入、输出来确定执行顺序和依赖关系,snakemake 每次默认执行第一个规则之后,会通过该规则的依赖关系(输入)来决定接下来执行哪个规则。所以,为了将所有规则都调用起来,我们需要在工作流文件最开始写下第一个规则:all 规则,来调取各支线最后一条规则的输出,这样层层递推,就可以将所有关联规则都调用起来。示例如下:
rule all:
input:
"stat/stat.txt"
12
生成可视化流程图
Snakemake 可以将整个工作流以流程图的形式导出(结合 dot 命令),文件格式可以使png、pdf等。命令如下:
$snakemake --dag -s test.py| dot -Tpdf > test.pdf
Building DAG of jobs...
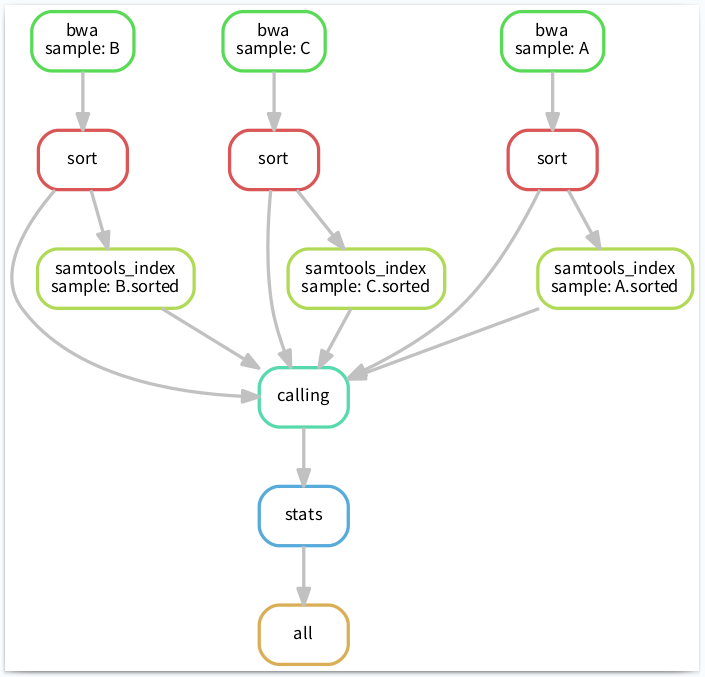
13
完整工作流
samples = [ "A", "B", "C"]
rule all:
input:
"stat/stat.txt"
rule bwa:
input:
"data/genome.fa",
"data/samples/{sample}.fastq"
output:
"mapped/{sample}.bam"
shell:
"bwa mem {input} | samtools view -Sb - > {output}"
rule sort:
input:
"mapped/{sample}.bam"
output:
"mapped/{sample}.sorted.bam"
shell:
"samtools sort -o {output} {input}"
rule samtools_index:
input:
"mapped/{sample}.bam"
output:
"mapped/{sample}.bam.bai"
shell:
"samtools index {input}"
rule calling:
input:
fa= "data/genome.fa",
bam=expand( "mapped/{sample}.sorted.bam", sample=samples),
bai=expand( "mapped/{sample}.sorted.bam.bai", sample=samples)
output:
"calling/all.vcf"
shell:
"samtools mpileup -g -f {input.fa} {input.bam} | "
"bcftools call -mv - > {output}"
rule stats:
input:
"calling/all.vcf"
output:
"stat/stat.txt"
:
"s/stat.py"
文:支永强
参考资料:
1. snakemake官方文档 https://snakemake.readthedocs.io/en/stable/
2.snakemake github https://snakemake.github.io
3. Snakemake技术手册https://snakemake.readthedocs.io/en/stable/tutorial/tutorial.html#tutorial
4. Snakemake常见问题https://snakemake.readthedocs.io/en/stable/project_info/faq.html#project-info-faq
5. Snakemake进阶https://snakemake.readthedocs.io/en/latest/executor_tutorial/tutorial.html#executor-tutorial
- 本文固定链接: https://maimengkong.com/zu/1348.html
- 转载请注明: : 萌小白 2023年1月15日 于 卖萌控的博客 发表
- 百度已收录
