
蛋白质组学(proteomics)专注于研究细胞、组织或整个生物体中的所有蛋白质,不仅关注蛋白质的存在,更重要的是它们的表达水平、修饰状态、相互作用以及功能。蛋白质组学的研究具有重要的科学和应用价值。以下,我们将通过一系列精心设计的问题,带您走进蛋白质组学的世界!
学习大纲
一、蛋白质组学研究意义何在?
二、市面上常见的蛋白质组学技术分类有哪些?
三、如何选择蛋白质组学技术?
四、蛋白质组学究竟如何做的?
五、蛋白质组学如何定性定量?
六、蛋白质组学采集方法选择?
七、蛋白质组学如何进行数据预处理和差异分析?
让我们跟着这些问题一起开启这段探索之旅吧!
Part.1、蛋白质组学研究意义何在?
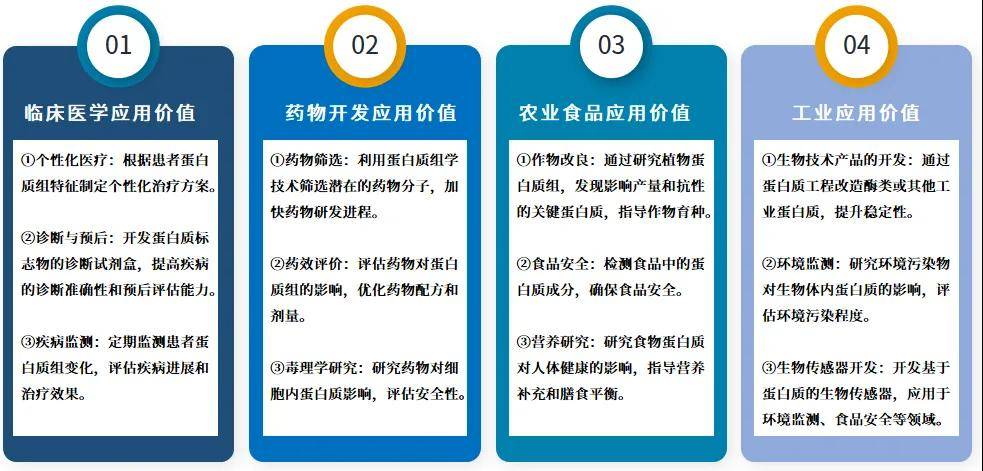
生物学基础科学研究
①蛋白质功能探索:蛋白质是生命活动的关键执行者,通过蛋白质组学研究可以发现新蛋白质的功能,从而更好地理解细胞过程、信号传导路径和代谢网络。
②蛋白质相互作用网络:研究蛋白质之间的相互作用有助于揭示内部复杂调控机制。
③翻译后修饰研究:探讨不同类型翻译后修饰及其对蛋白质功能的影响。
疾病机理科学研究
①疾病标志物的发现:通过比较不同个体的蛋白质组差异发现新的疾病标志物,有助于疾病的早期诊断。
②药物靶点鉴定:识别潜在的药物作用靶点,为药物研发提供科学依据。
③疾病机制解析:通过蛋白质组学研究可以揭示疾病的分子机制,帮助理解疾病的发生和发展过程。
Part.2、市面上常见的蛋白质组学技术分类有哪些?
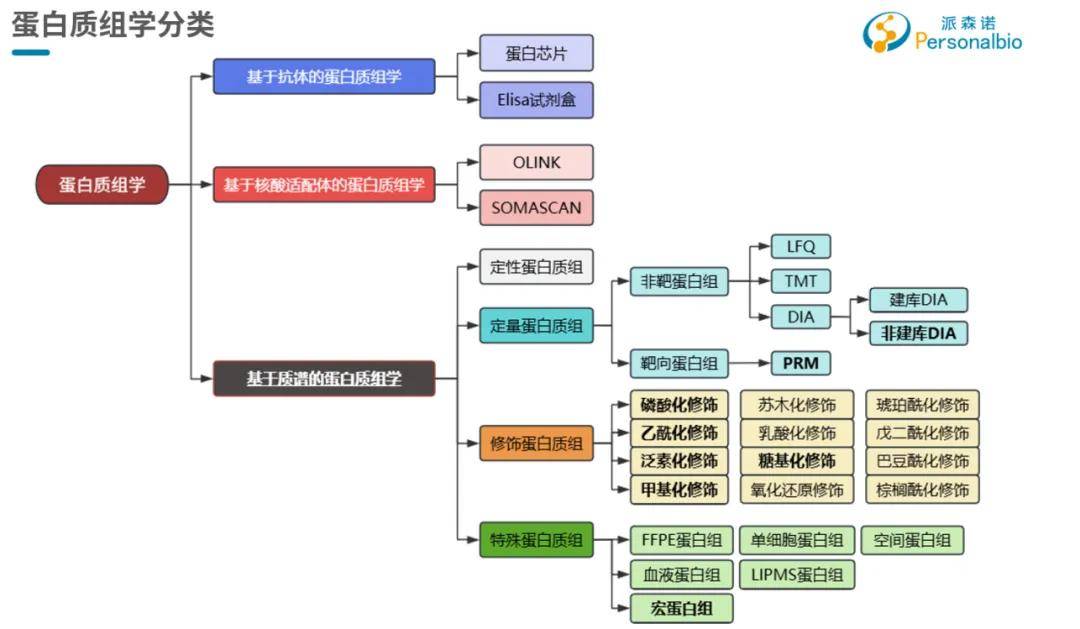
根据研究方法和技术的不同,蛋白质组学可以分为多个子领域或分类。这些分类不仅反映了不同的检测原理和技术手段,还体现了研究者们针对特定科学问题所采取的不同策略。例如根据检测原理的不同可将蛋白质组学大致分为三类。
1. 基于抗体的蛋白质组学:利用特异性抗体来检测和量化蛋白质,主要原理是抗原抗体结合反应。
2.基于核酸适配体的蛋白质组学:利用核酸适配体(Aptamers)来检测和量化蛋白质,核酸适配体是一种具有高亲和力和特异性的小分子核酸片段,可以与特定的目标分子结合,由于蛋白不能像核酸一样扩增,因此该技术针对特定的蛋白质设计相对应的核酸适配体,与蛋白质结合,从而实现将蛋白质信号转变成核酸信号进行检测。
3.基于质谱的蛋白质组学:目前最常用且最具代表性的蛋白质组学方法之一,通过质谱仪分析肽段的质量和电荷比(m/z),从而识别和量化蛋白质。
Part.3、如何选择蛋白质组学技术?
那么市面上常见的方法如此多样,他们究竟适用于什么情况?小编用一张图总结如下。
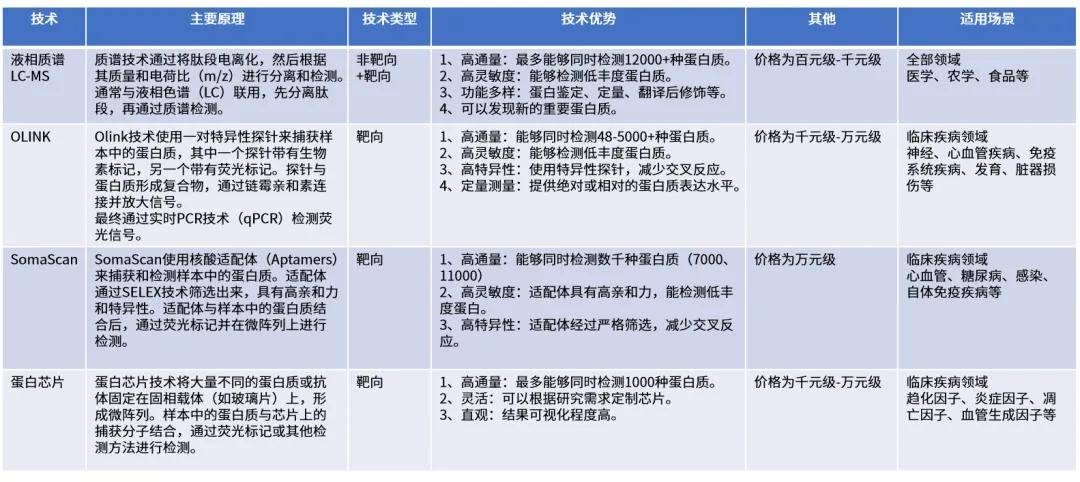
SomaScan、Olink、蛋白芯片和质谱技术各有优势和局限性,选择哪种技术取决于具体的研究需求、样本类型、研究目的和预算等因素。在实际应用中,可能会结合多种技术以获得更全面和准确的数据,也起到了互为补充的作用。
Part.4
蛋白质组学究竟如何做的?
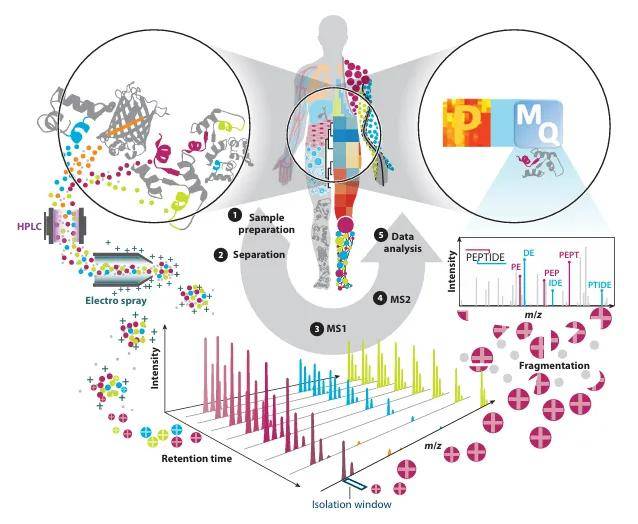
接下来我们将从蛋白质组学流程进行介绍,帮助大家对基于质谱的蛋白质组学有更清楚的了解。
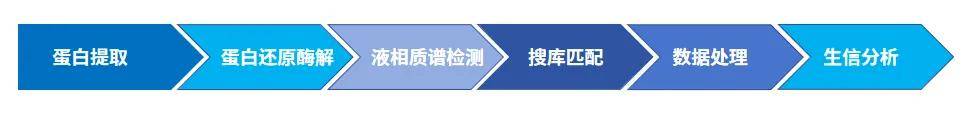
蛋白提取
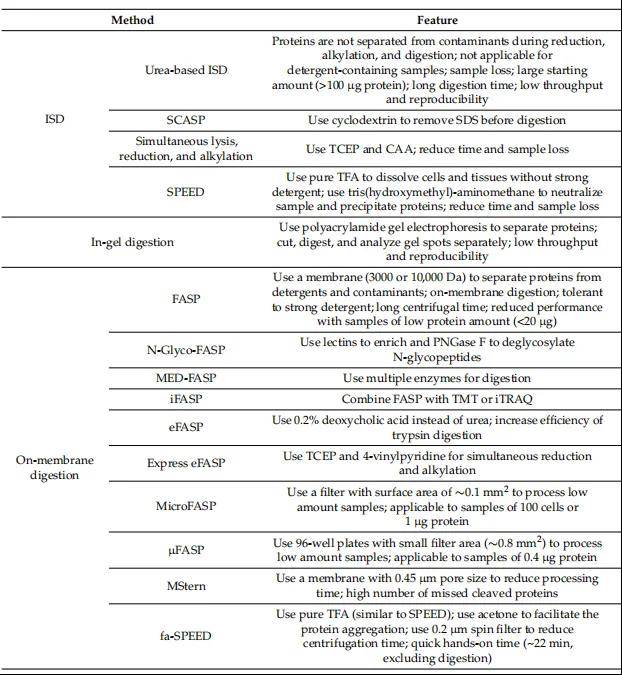
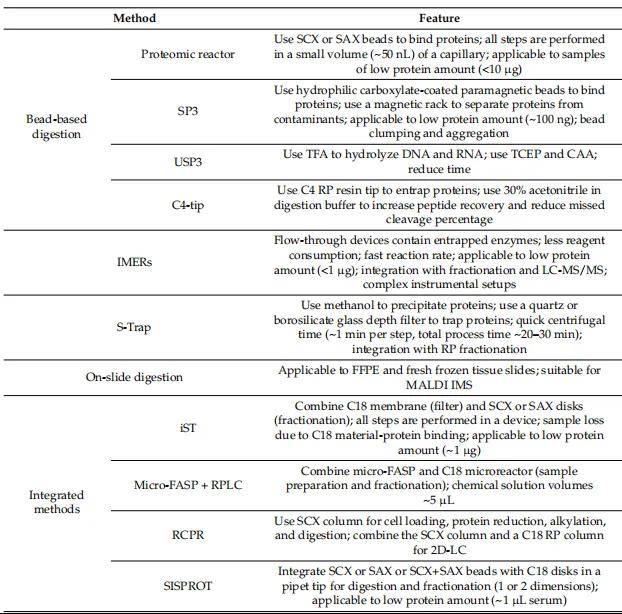
蛋白质组学样品制备方法包括从生物基质中提取蛋白质,去除非蛋白质污染物,如DNA、糖和脂类,去除电离过程中可能形成加合物的残留盐,以及蛋白质分离以降低样品的复杂性。蛋白质提取方法的选择取决于样品类型(如细胞、组织、血液等)、研究目的以及所需蛋白质的性质。目前主流的蛋白提取方法包括机械破碎(匀浆、研磨)、物理破碎(超声、温差)、化学破碎(变性剂、去垢剂),在整个提取过程中保证低温环境,以及添加蛋白酶抑制剂防止蛋白降解。根据样本的不同,有时对于低丰度蛋白质或者修饰蛋白质的富集是一个关键的步骤。对于去除可能来自生物样品或在提取/耗尽/富集中添加的小分子,有许多方法可用,包括透析、缓冲液交换、尺寸排除、蛋白质沉淀、色谱或电泳。在所有的脱盐方法中,用有机溶剂(丙酮或甲醇/氯仿)沉淀是最便宜、最简单、最可扩展的脱盐蛋白质的选择。
蛋白还原酶解
使用还原剂将蛋白质中的二硫键打断,其中DTT (Dithiothreitol)和TCEP (Tris(2-carboxyethyl)phosphine)是常用的还原剂,能够有效地打断二硫键。紧接着为了保护还原后的巯基进行烷基化步骤,避免它们重新形成二硫键或被氧化,碘乙酰胺 (Iodoacetamide,IAM)是最常用的烷基化试剂,烷基化剂通常会封闭半胱氨酸的巯基,使其不再参与氧化反应。在此基础上通常通过添加胰蛋白酶将蛋白酶解成肽段以备上机。
液相质谱检测
肽段在纳升级液相系统可实现复杂肽混合物的显著分离,其中分离时间和流动相的pH是纳升级液相质谱技术的关键影响因素。当肽段经过液相分离后,进入质谱系统进行检测。质谱系统主要由进样系统、离子源、质量分析器、检测器构成。
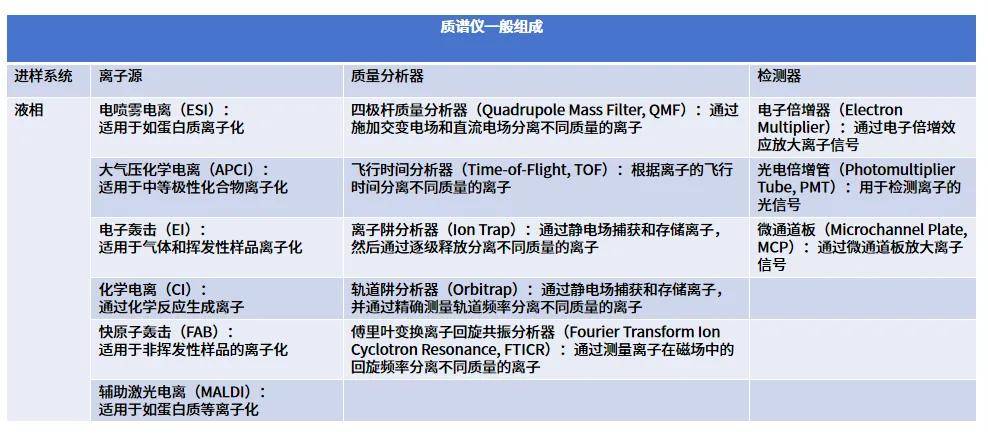
值得一提的是质谱系统电喷雾电离(Electrospray Ionization, ESI)能够将液态样品转化为带电的气态离子,广泛用于蛋白质组学。肽段溶液通过一根细长的毛细管喷出,液滴迅速蒸发,体积逐渐减小,表面的电荷密度增加到一定阈值时,库仑斥力超过了表面张力,液滴会分裂成纳米级的具有极高电荷密度的液滴,带电离子进入质谱仪检测。
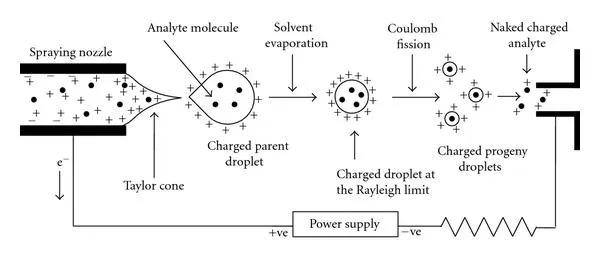
离子在质量分析器中被引入电场,按照质荷比(m/z)进行分离。离子在电场中振荡,产生频率信号。频率信号被转换为质谱图,显示出各离子的m/z值及其强度,此时获得的质谱图称为一级质谱,此时的检测的肽段称为母离子。若想得到更准确的信息则需从一级质谱中选择母离子,通过碰撞诱导裂解等方式使母离子断裂成子离子,记录子离子的m/z值和强度信息。具体过程见以下动画:
,时长
01:06
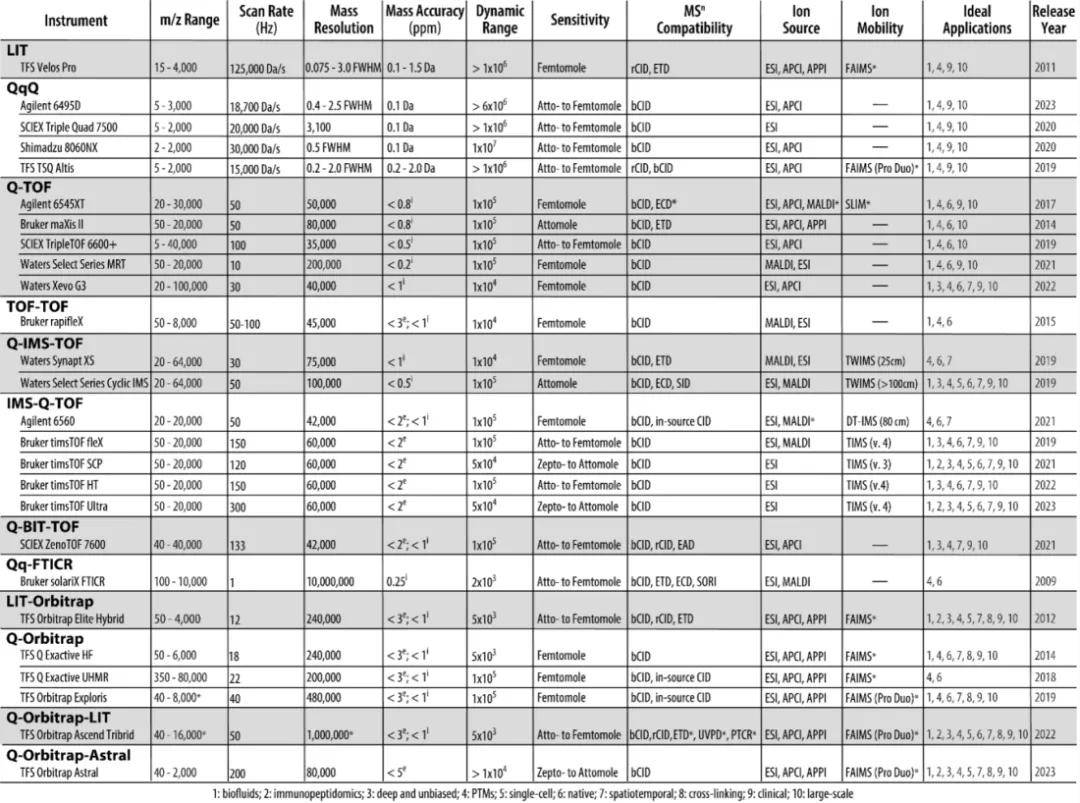
补充知识:业内通常把分辨率在10000(FWHM)以上的质谱称为高分辨质谱(分辨率即是指质谱仪区分两个质量相近的离子的能力),主要包括飞行时间质谱(TOF MS)、轨道阱质谱(Orbitrap MS)及傅里叶变换离子回旋共振质谱(FT-ICR MS)。在高分辨质谱领域中,商业化应用最成功的产品当数Thermo的Orbitrap轨道阱质谱系列及Agilent、Waters、Bruker的飞行时间质谱系列。其中Thermo的Astral质谱仪表现尤为优异,其检测能力在我们之前的测试数据中可以窥见。(震撼登场!质谱黑科技——Astral,引领未来分析新潮流!点击查看)
Part.5、蛋白质组学如何定性定量?
离子化后的肽段(通常称为母离子)进入质量分析器后,由于各自的质荷比不同,在电场或磁场中的运动轨迹不同,因此可以通过检测器的位置和时间来计算离子的质荷比(m/z)。最后形成一张张质荷比从左到右依次增大的谱图,也就是一级质谱图,一级质谱中每一个母离子都会有一个离子峰,离子峰的面积大小代表着母离子的丰度,也就是蛋白质的相对含量。因此,一级质谱可以帮助我们对蛋白进行定量分析。
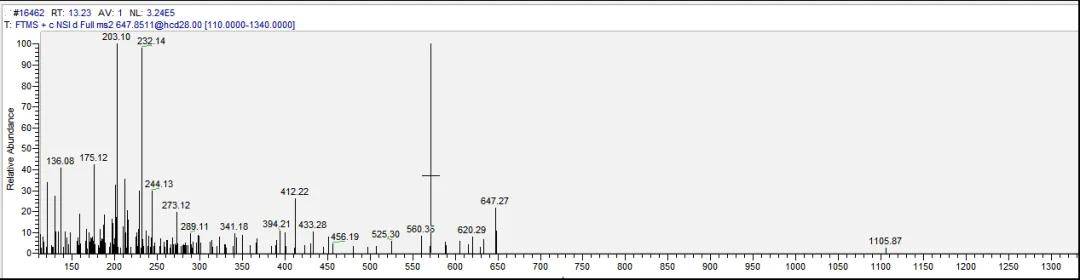
那么通过一级质谱了解到了蛋白质的相对含量,但这些蛋白质都是什么蛋白呢,这就需要二级质谱对蛋白质进行定性。
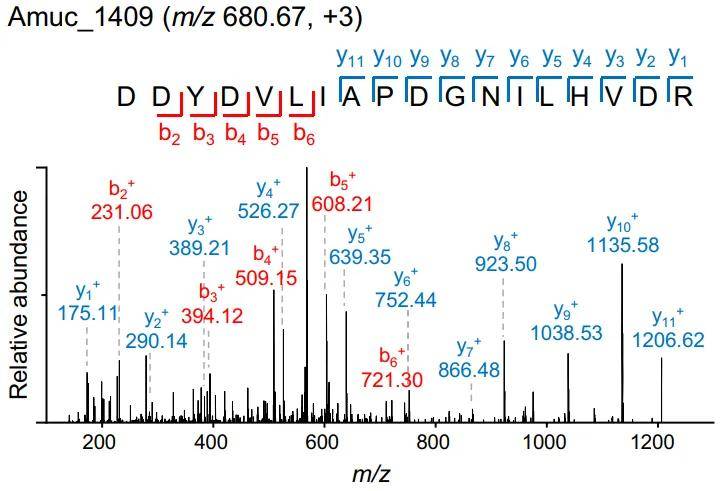
在二级质谱分析实验中,质谱仪会选择一级质谱中的一个峰,也就是对应质荷比的肽段离子(母离子),让这些离子高速撞击质谱仪中的惰性气体造成肽键随机断裂,通过分析肽键断裂产生的二级质谱图即可获得被选择碰撞的肽段序列。相比起一级质谱仅测得多肽的质量,二级质谱能够鉴定多肽的序列,比一级质谱获得的信息更为精准详细。
肽键二级断裂的方式有非常多种可能性,通常质谱撞击使用的电压较低,其他系列的离子不易产生,因此产生的离子类型通常考虑b离子系列和y离子系列(by离子是二级质谱中常见的碎片离子类型,它们是肽段裂解后产生的两种主要类型的子离子,b离子是从肽段的N端开始断裂产生的离子,y离子是从肽段的C端开始断裂产生的离子,b离子和y离子是互补的,即它们共同覆盖了整个肽段的氨基酸序列,检测b离子和y离子的m/z值及其强度信息,可以推断出肽段的氨基酸序列。也可以检测肽段上的翻译后修饰)。
获取到蛋白质组学质谱原始数据后便要进行数据库检索,该过程是将质谱仪采集到的原始谱图数据与理论蛋白质组数据库中的理论谱图进行比对。蛋白质组数据库通常包含大量已知蛋白质的序列信息,通过模拟酶切和碎裂过程,可以将这些序列信息转换为理论谱图数据。(关于蛋白质数据库的选择问题,小编之前也有过详细介绍,建议优先使用Uniprot数据库,其他情况可以查阅链接-资源分享,蛋白质研究常用数据库点击查看)
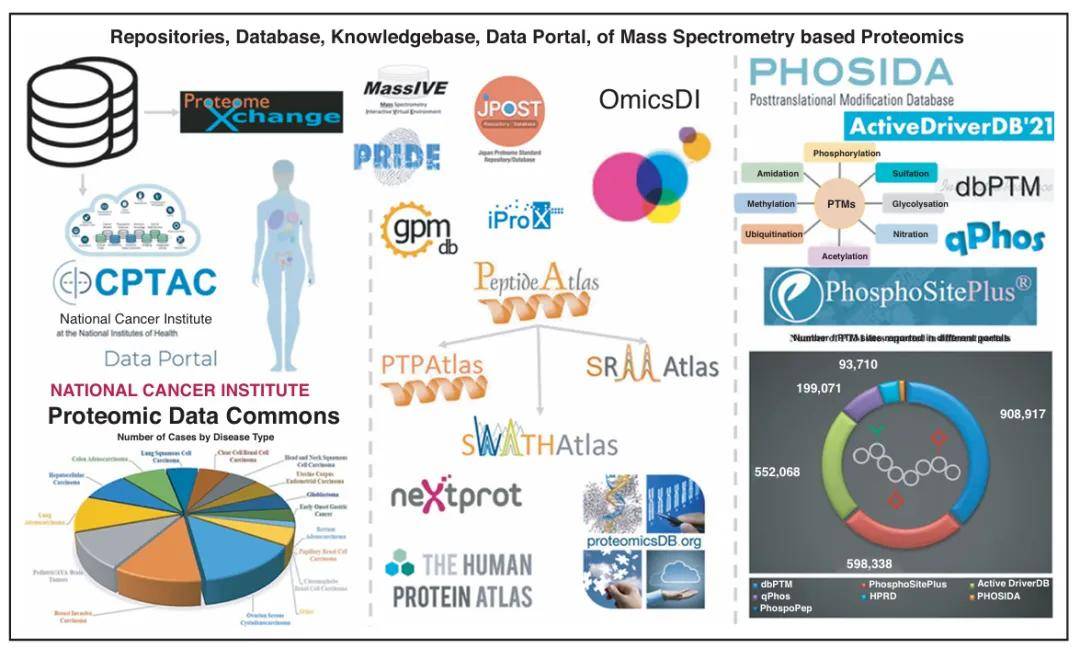
然后,利用计算机软件对原始谱图数据进行解析,提取特征信息,并与理论谱图进行匹配,从而推断出样品中存在的蛋白质种类及其相对丰度。目前已有多种商业和开源的搜库软件可供选择,如Proteome Discoverer、MaxQuant等。这些软件通常具有强大的数据处理和分析能力,能够支持多种质谱仪产生的原始数据格式,并提供丰富的后处理功能。
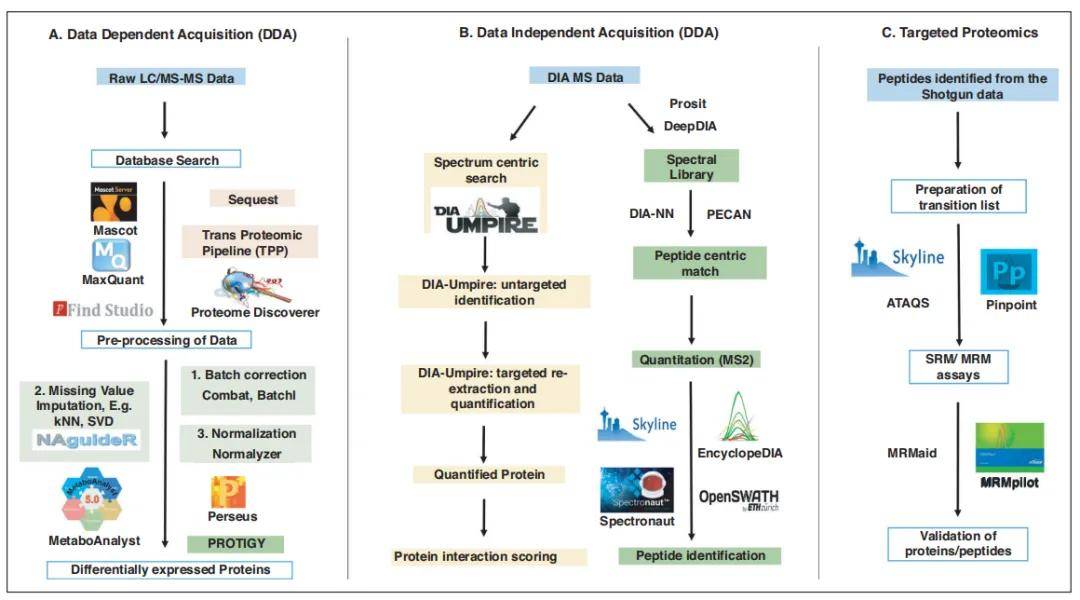
Part.6、蛋白质组学采集方法选择?
挑选一级母离子丰度靠前的肽段碎裂的方式为DDA-数据依赖型采集;不依赖于一级母离子丰度而是分窗口按照m/z范围进行二级碎裂的方式叫DIA-数据非依赖型采集。各类方法的选择和技术比较可阅读之前的公众号推文(必看!!!非标记蛋白质组学哪“家”强?点击查看)
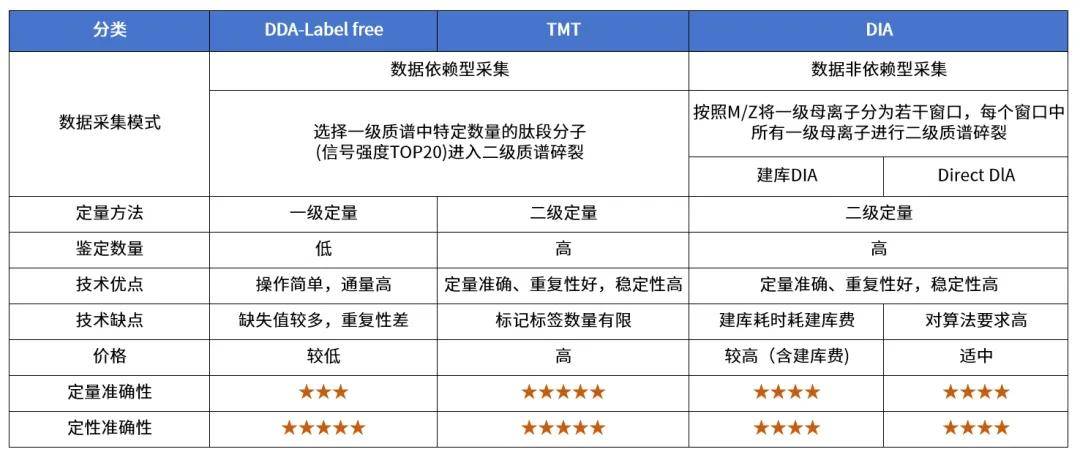
Part.7、蛋白质组学如何
进行数据预处理和差异分析?
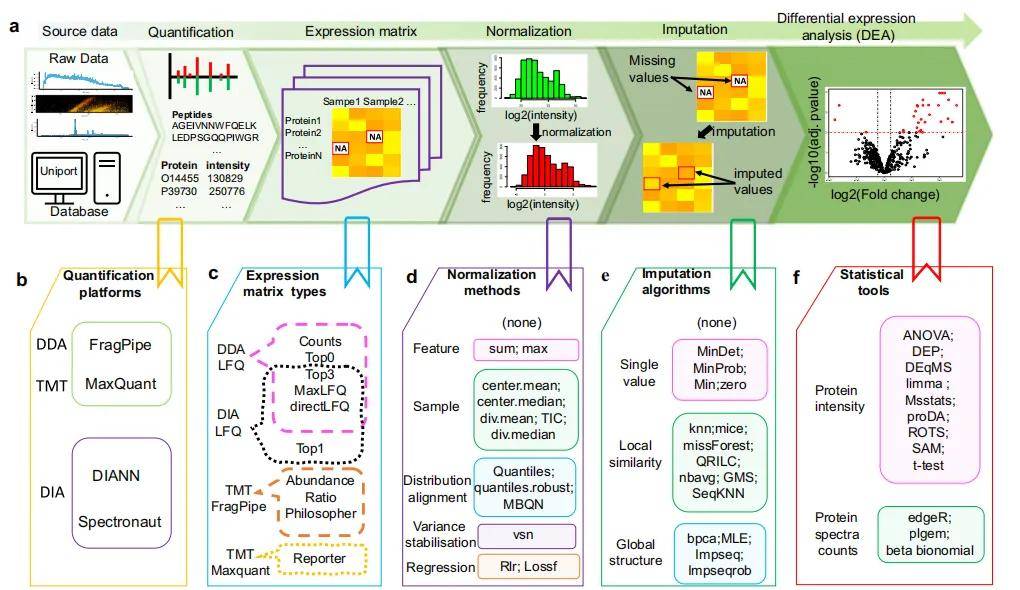
蛋白质组学数据预处理是数据分析流程中至关重要的步骤,直接影响到后续分析的质量和准确性。预处理的步骤包括数据清理、归一化、缺失值填补、批次效应校正等。
数据清理 (Data Cleaning)
去除低置信度数据:通常根据质谱分析中FDR (False Discovery Rate) 控制。
去除污染物和背景蛋白:使用MaxQuant去除样品中的外源性蛋白。
保留生物重复信息:如果进行差异蛋白分析,生物重复数据可以用于评估统计显著性。
缺失值填补 (Missing Value Imputation)
缺失值处理是蛋白质组学数据的核心问题之一。以下是几种常见的填补方法:
删除缺失值:如果某些蛋白质在大多数样本中未被检测到,考虑将这些数据删除。
零填补:直接将缺失值填补为零,但这种方式可能导致误差增加。
KNN填补(K-Nearest Neighbors):基于数据集中的相似样本来填补缺失值。
基于均值/中位数填补:用全体样本的均值或中位数填补缺失值。
随机森林填补:通过构建随机森林模型来预测缺失值。
数据归一化 (Normalization)
归一化是为了解决不同样本之间的技术变异,确保定量结果具有可比性。常用的归一化方法有:
总强度归一化 (Total Ion Current, TIC Normalization):假设所有样本中总蛋白质含量相同,将每个样本的蛋白质丰度按总强度进行归一化。
分位数归一化 (Quantile Normalization):通过将所有样本的强度分布调整为相同的分布,消除样本之间的变异。
中值归一化 (Median Normalization):对每个样本进行中值归一化,平衡不同样本之间的差异。
批次效应校正 (Batch Effect Correction)
蛋白质组学实验涉及多个批次的实验操作时需要校正批次效应以避免批次间的技术变异影响分析结果。常用的批次效应校正方法有:
ComBat:一种基于经验贝叶斯方法的批次效应校正方法,广泛应用于基因组和蛋白质组学数据。
SVA (Surrogate Variable Analysis):通过构建伪变量来校正隐藏的批次效应。
差异分析 (Differential Expression Analysis)
Student's t-test:用于假设方差相等的两组之间的差异。
方差分析 (ANOVA):当有多个实验组时,ANOVA用于比较多个组之间的差异。
非参数检验:如果数据不符合正态分布可以使用非参数检验,如Kruskal-Wallis检验等。
小结:本期内容介绍了蛋白质组学技术情况、样本准备、仪器定性定量原理和蛋白质组学搜库软件以及蛋白质组学数据预处理,且下游分析丰富多样。如何应用这些分析找到关键蛋白以及如何进行后续研究,或许是不少老师的困扰,小编将持续为大家带来系列分享,敬请期待哦!
参考文献
1、Duong VA, Lee H. Bottom-Up Proteomics: Advancements in Sample Preparation.Int J Mol Sci. 2023;24(6):5350. Published 2023 Mar 10.
2、Peters-Clarke TM, Coon JJ, Riley NM. Instrumentation at the Leading Edge of Proteomics. Anal Chem. 2024;96(20):7976-8010.
3、Halder A, Verma A, Biswas D, Srivastava S. Recent advances in mass-spectrometry based proteomics software, tools and databases.Drug Discov Today Technol. 2021;39:69-79.
4、Halder A, Verma A, Biswas D, Srivastava S. Recent advances in mass-spectrometry based proteomics software, tools and databases.Drug Discov Today Technol. 2021;39:69-79.
5、Peng H, Wang H, Kong W, Li J, Goh WWB. Optimizing differential expression analysis for proteomics data via high-performing rules and ensemble inference.Nat Commun. 2024;15(1):3922. Published 2024 May 9. doi:10.1038/s41467-024-47899-w.
- 本文固定链接: https://maimengkong.com/zu/1855.html
- 转载请注明: : 萌小白 2024年12月13日 于 卖萌控的博客 发表
- 百度已收录
