Samtools作为一款操作序列比对结果文件(SAM/BAM)的工具,能够灵活转换sam/bam,并且能基于参考序列和Sam/bam文件进行变异位点检测,并通过bcftools进行变异结果统计。本期将基于上期bwa软件的比对结果,利用samtools进行变异检测。
一、软件安装
-
下载地址http://www.htslib.org/download/,利用wget下载得到samtools-1.5.tar.bz2,bcftools-1.5.tar.bz2;
-
解压缩:tar -jxvf bcftools-1.5.tar.bz2
tar -jxvf samtools-1.5.tar.bz2
-
安装(详细的安装信息可查看bcftools-1.5(samtools-1.5)文件夹下的INSTALL):
cd bcftools-1.5 ( samtools-1.5)
./configure –prefix=/where/to/install#设置安装路径
Make
Make install
-
安装完成后,我们后面所用到的可执行程序都在上面设置的安装路径(/where/to/install)的bin文件夹下;
-
配置环境变量
a.若要临时修改环境变量,可直接在终端输入下面一行命令:
Export PATH=/where/to/install/bin:$PATH
b.要永久修改环境变量可将下面第一行添加到~/.bash_profile(针对当前用户)或者/etc/profile(针对所有用户)文件的末尾,再执行第二行命令即可:
Export PATH=$PATH: /where/to/install/bin;
source ~.bash_profile 或者source /etc/profile
现在samtools 和bcftools已经安装好了,下面我们就进行calling SNPs和Indels了。
二、使用流程
1. 输入文件
比对结果文件sample.sort..bam和参考基因组序列ref.fa(上一期已对其建立索引,此处不再建立索引);
2. Variant Calling主要流程
Samtools index sample.sort.bam#对bam文件建立索引,默认生成文件为bam文件加.bai,此处生成sample.sort.bam.bai;
Samtools rmdup sample.sort.bam sample.dedup.bam#去除PCR等实验过程中产生的多余duplications;
Samtools mpileup –q 20 –d 8000 –ugo sample.bcf –f ref.fa sample.dedup.bam#variant calls
参数说明:
-q, --min-MQ mapQ最小值,低于这个值则跳过;
-d, --max-depth 最大覆盖深度;
-g, --BCF 生成bcf格式文件;
-o, --output FILE 输出文件;
-f , --fasta-ref FILE参考序列(fasta format)的索引文件名;
其他常用参数:
-A --count-orphans 不丢弃异常配对reads;
-l --positions FILE 包含区域位点的位置列表文件或者bed区域文件;
-r --region REG 在指定区域产生pipeup,和-l一起使用;
-I --skip-indel 不检测INDEL;
-m --min-ireads 候选INDEL的最小间隔的reads数;
-v --VCF 生成VCF格式文件;
-u --uncompressed 产生未压缩的vcf/bcf文件;
Bcftools call –vmO z –o sample.vcf.gz sample.bcf #将bcf转化为vcf文件,bcf为vcf的二进制格式
参数说明:
-v --variants-only 仅输出变异位点,不加此参数,则输入所有位点信息;
-c --consensus-caller the original calling method;
-m -- multiallelic-caller alternative model for multiallelic and rare-variant calling,calling的方法,与-c不能同时使用;
-O --output-type <b|u|z|v> 指定输出文件类型, 'b'表示压缩后BCF文件; 'u'表示未压缩的BCF文件; 'z'表示压缩的VCF文件; 'v' 表示未压缩的VCF文件;
三、vcf文件的过滤统计
bcftools filter -O z -o sample.filtered.vcf.gz -i 'QUAL>20 && DP>5' sample.vcf.gz#过滤质量值低于20并且深度小于5的变异位点
参数说明:
-O --output-type <b|u|z|v> 指定输出文件类型, 'b'表示压缩后BCF文件; 'u'表示未压缩的BCF文件; 'z'表示压缩的VCF文件; 'v' 表示未压缩的VCF文件;
-o, --output FILE 输出文件;
-i --Include <exp> 筛选出满足exp条件的变异位点;
-e --exclude<exp> 剔除满足exp条件的变异位点,与-i相反;
--threads 线程数;
Note: 上面的过滤条件中可以加入DP4参数,过滤出高质量的变异位点
Vcf 文件格式:由#开头的注释部分(图1)和变异主体部分(图2,图3),注释部分主要包括软件版本,命令行,参考序列信息以及主体部分参数的注释,主体部分由以tab隔开的8列组成,依次为参考序列名,variant的位置,variant的ID,参考序列的碱基,Phred格式(Phred_scaled)的质量值,过滤结果和第8列variant 的详细信息(图3),采用tag=value形式,tag间用“;”隔开;
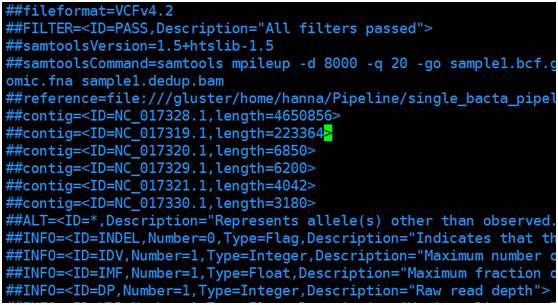
图1 VCF文件注释部分

图2 VCF文件变异主体部分前7列
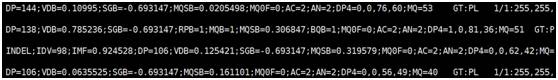
图3 VCF文件变异主体部分详细信息和基因型列
bcftools stats sample.filtered.vcf.gz > sample.stat.file# 统计SNPs和Indels变异数目等,包括转换,颠换数目;
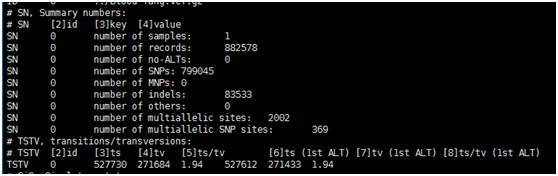
sample.stat.file部分内容如下图所示,number of SNPs为SNPs总数目,number of indels为发生indels的总数目,ts表示转换数目,tv表示颠换数目:
bcftools view –v snps –g hom sample.filtered.vcf.gz# 仅输出纯合SNPs,通过改变参数可以输出杂合SNPs,纯合/杂合Indels;
参数说明:
-h 仅输出头部注释文件;
-H 不输出头部注释文件;
-O --output-type <b|u|z|v> 指定输出文件类型, 'b'表示压缩后BCF文件; 'u'表示未压缩的BCF文件; 'z'表示压缩的VCF文件; 'v' 表示未压缩的VCF文件;
-T, --threads 设置线程数目
-i --Include <exp> 筛选出满足exp条件的变异位点;
-e --exclude<exp> 剔除满足exp条件的变异位点,与-i相反;
-v --types [snps|indels|mps|other],仅输出指定类型的变异位点信息;
-V --exclude-type [snps|indels|mps|other],仅输出不包含指定类型的变异位点信息;
-g --genotype [hom|het|miss],仅输出指定基因型的位点。
四、参考
Samtools官方说明文档:http://www.htslib.org/doc/#manual-pages
Bcftools官方说明文档:http://samtools.github.io/bcftools/bcftools.html
参考文献:
-
Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. and 1000 Genome Project Data Processing Subgroup (2009)The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics, 25, 2078-9. [PMID: 19505943]
-
Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011 Nov 1;27(21):2987-93. Epub 2011 Sep 8. [PMID: 21903627]doi:10.1093/bioinformatics/btr509;
-
Danecek P., Schiffels S., Durbin R. Multiallelic calling model in bcftools (-m) [link]
-
Li H. Improving SNP discovery by base alignment quality. Bioinformatics. 2011 Apr 15;27(8):1157-8. doi: 10.1093/bioinformatics/btr076. Epub 2011 Feb 13. [PMID: 21320865]
-
Durbin R. Segregation based metric for variant call QC [link]
-
Li H, Mathematical Notes on SAMtools Algorithms [link]
-
The Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes[J]. Nature, 2012, 491(7422):56-65. doi: 10.1038/nature11632
-
Wu Y, Jing R, Dong Y, et al. Functional annotation of sixty-five type-2 diabetes risk SNPs and its application in risk prediction[J]. Scientific Reports, 2017, 7.
- 本文固定链接: https://maimengkong.com/zu/1347.html
- 转载请注明: : 萌小白 2023年1月15日 于 卖萌控的博客 发表
- 百度已收录
