一. 常规BSA
QTL-seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations(The Plant Journal)
BSA(Bulk segregation analysis)进行数量性状基因定位的方法由来已久,但是将高通量测序应用在BSA上则是近年来才出现的方法。这篇水稻的文章是首次将高通量测序应用在BSA分析上的尝试,之后进行的常规BSA测序基本沿用该文献提供的方法。
本文创新地将高通量测序和传统的BSA方法结合在一起,并且对分析方法进行了详细的描述。
使用两个性状差异较大的亲本A和B构建遗传群体(如图示为水稻植株高矮性状,构建的F2群体),由于多个数量性状位点控制植株的高矮,该性状在F2群体中呈现正态分布。从群体中选取最高的多个植株和最矮的多个植株分别提取DNA组成“高”“矮”混池,并且将两个混池分别进行高通量测序。测序完成后将测序数据比对到A亲本水稻的参考基因组上,来进行SNP-Index的计算。
SNP-Index是基于高通量测序的BSA分析中最重要的概念,简而言之是位点上与某个亲本一致的reads占总reads的比例,可以看成子代混池中某种等位基因的频率。以下图为例,10条reads覆盖了某一个核酸位点,该位点的测序深度是10。在这10条reads当中,如果有4条reads包含与参考基因组不同的碱基,则SNP-Index为0.4;如果10条reads均包含与参考序列不同的碱基,则SNP-Index为1。
将两个极端混池的SNP-Index相减得到ΔSNP-Index。通过滑窗计算染色体上各个区域的ΔSNP-Index,可以判断染色体上哪个区域最有可能包含目标位点。
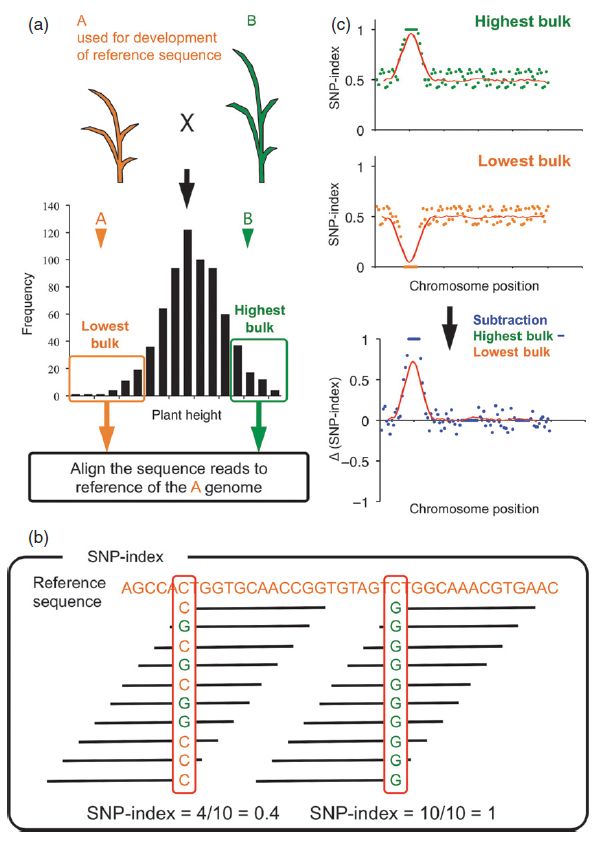
图1 基于高通量测序的BSA分析示意图
研究者利用这种方法进行了水稻抗稻瘟病的性状定位。使用较抗病的Nortai和易感病的Hitomebore两个品系作为亲本,构建了重组自交系群体(RIL)。对子代群体中的个体使用稻瘟病菌处理2周,选取较抗病和易感病个体分别构成混池,进行测序。对测序数据进行SNP-Index分析,发现在6号染色体上2.39-4.39 Mb的位置ΔSNP-Index最高,最有可能包含目标性状基因。
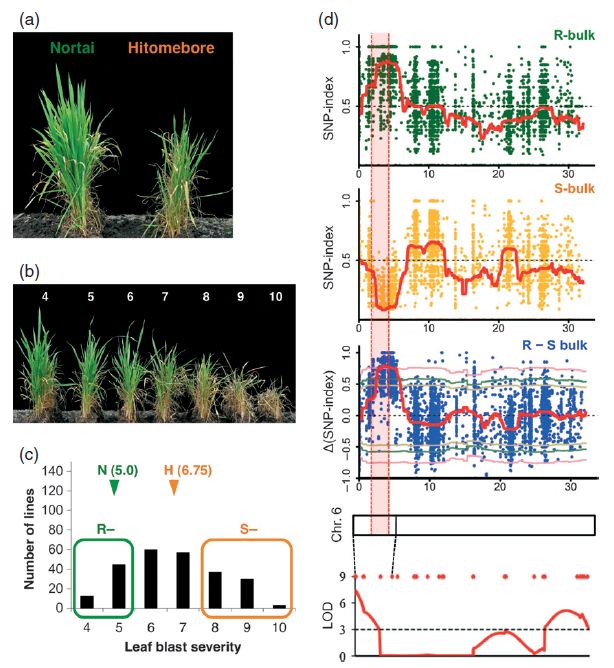
图2 水稻抗稻瘟病性状定位结果
研究者还对水稻幼苗活力性状进行了定位。使用幼苗活力较高的Dunghan Shali和幼苗活力较低的Hitomebore品系杂交构建F2代分离群体,在种子吸胀后第14天测定幼苗高度,选取最高的多个个体与最矮的多个个体分别构建混池,并进行高通量测序。SNP-Index分析发现在3号染色体的36.21-37.31 Mb处和1号染色体的39.08-41.08 Mb处最可能包含目标性状位点。
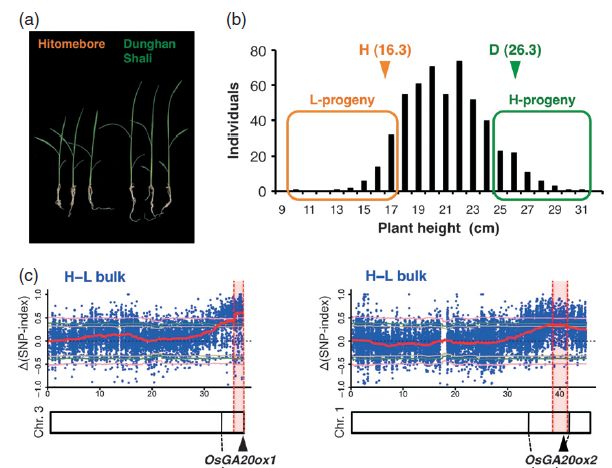
图3 水稻幼苗活力性状定位结果
二. Mutmap
Genome sequencing reveals agronomically important loci in rice using MutMap(Nture Biotechnology)
Mutmap是BSA分析的一个重要变种,其基本原理也是通过构建分离群体,挑选极端性状构建混池,并通过计算SNP-Index来对目标性状位点进行定位。但是Mutmap可以针对经过化学诱变的个体进行性状定位,在对诱变得到的性状进行定位的时候更具优势。
文章对Mutmap方法以及后续分析进行了细致的描述。
将野生型植株使用甲磺酸乙酯(ethyl methanesulfonate, EMS)进行诱变,得到包含目的性状的突变株后与野生型亲本回交,得到F1后自交产生F2代分离群体。从F2代中挑选野生型与突变型性状个体分别构建混池进行高通量测序。测序完成之后,同样是与参考基因组进行比对,并计算SNP-Index。
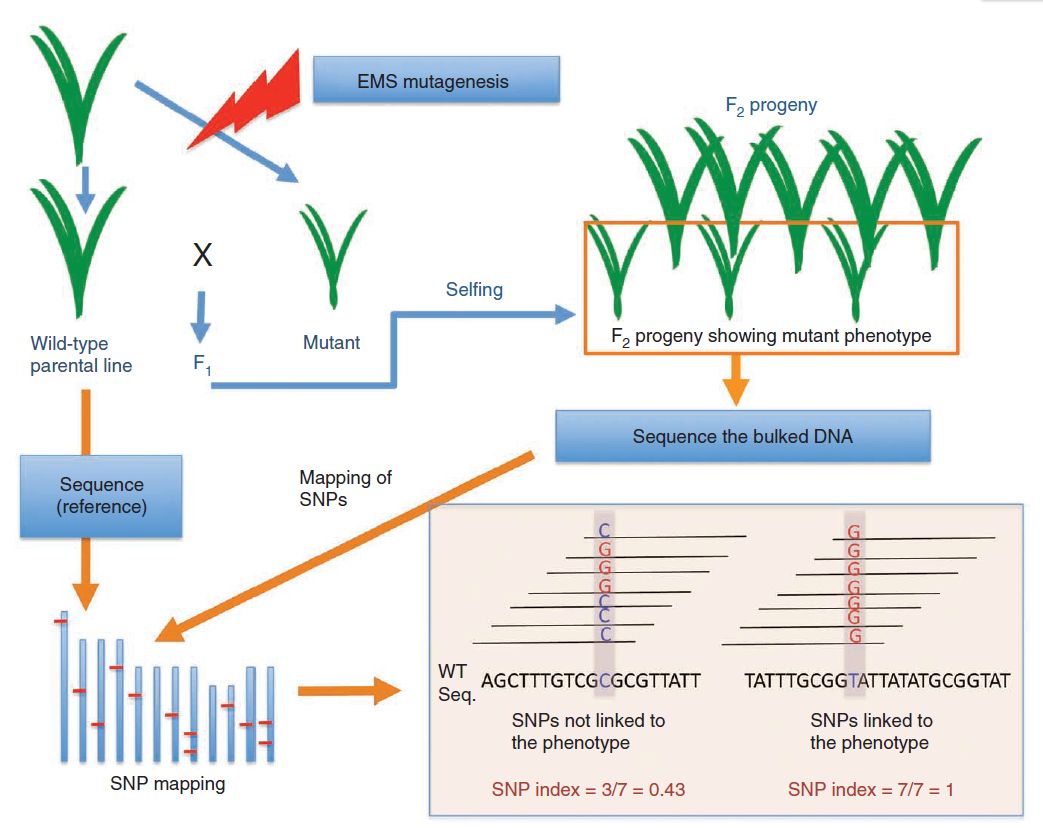
图4 Mutmap方法示意图
研究者利用建立的Mutmap方法对水稻叶片颜色性状进行了定位。以Hitomebore作为野生型使用EMS进行诱变,得到叶片颜色较浅的突变株Hit1917-pl1和Hit0813-pl2,并将这两株与亲本回交后分别进行了Mutmap定位。SNP-Index计算的结果将目标性状分别定位到10号染色体和1号染色体上。其中10号染色体的位置包含一个疑似目标基因OsCAO1。
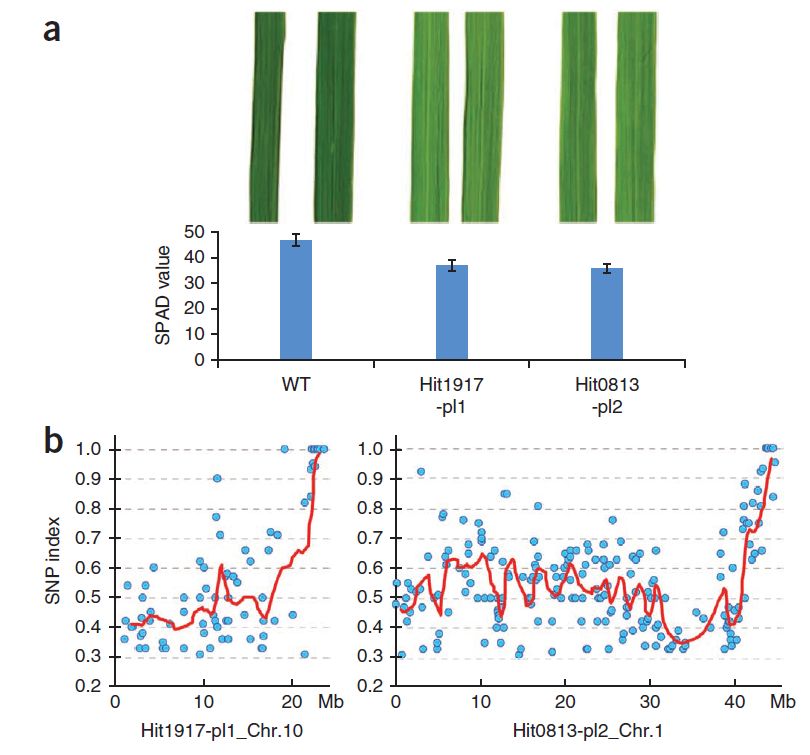
图5 水稻叶片颜色突变的定位结果
研究者使用转基因方法进行了验证,分别往突变株Hit1917-pl1中转入了OsCAO1,往野生型植株中转入了敲低OsCAO1表达量的RNA干扰。往突变株Hit1917-pl1中转入OsCAO1得到的植株叶片颜色变深,野生型植株敲低OsCAO1表达量后叶片颜色变浅,验证了OsCAO1基因对水稻叶片颜色的调控作用。
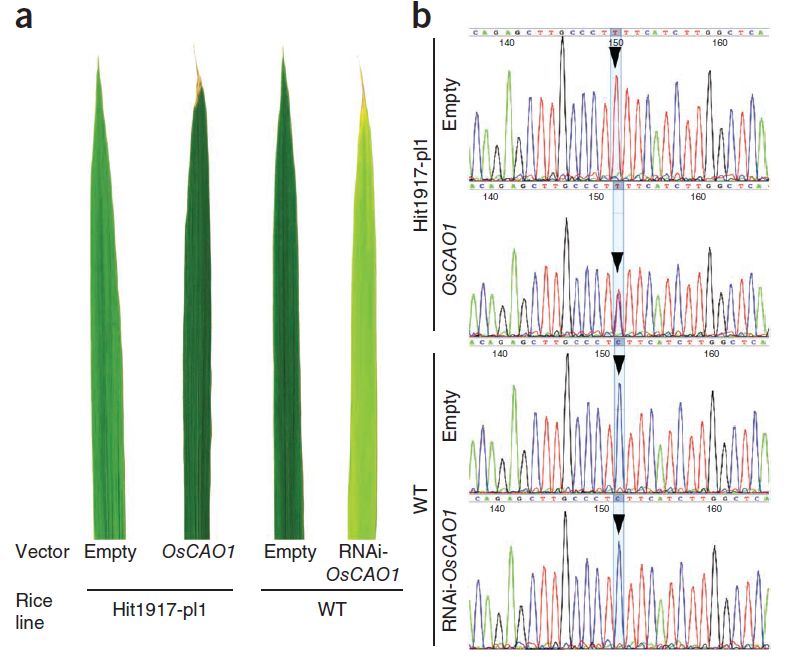
图6 对OsCAO1基因功能进行的转基因验证,以及OsCAO1基因cDNA的测序峰图
三. BSR
Gene mapping via bulked segregant RNA-seq (BSR-seq)(PloS ONE)
对于BSA分析来说,关注的突变位点更多地位于基因的编码区域,而且部分物种基因间区域包含大量重复序列,对测序和分析造成干扰。在这种情形下,使用RNA测序来进行BSR分析,不失为一个更好的选择。
BSR方法构建群体、选择混池的方式与常规的BSA方法并无二致,只是把测序对象从DNA更换成了RNA。研究者对玉米的gl3基因突变株进行了BSR分析,gl3基因会影响玉米叶片上蜡质的积累。研究者将gl3基因突变株与野生型作为亲本构建了F2代分离群体,从F2代中挑选极端性状构建了2个混池,并分别进行了RNA测序。将测序数据与参考基因组比对后进行SNP识别,并使用经典贝叶斯算法计算SNP与性状连锁的概率。最后将gl3基因定位到了4号染色体上一段长为2 Mb的位置上,并在该位置找到了gl3基因,该基因编码啊一个可能的myb转录因子。

图7 玉米BSR测序定位结果
作者提到,对于BSR测序来说,在处理大基因组,尤其是那些包含大量重复序列的基因组的时候会很有优势。不仅减少了数据的冗余,也降低了测序成本。同时大量的RNA测序数据,也可以通过表达量的变化来为候选基因的筛选提供相应的证据。但是同时值得注意的是,对于一些发生在调控区域的突变,BSR测序是无能为力的,可能存在与性状关联不上的风险。所以BSR虽然是一个很有力的研究工具,但使用的时候仍然需要谨慎。- 本文固定链接: https://maimengkong.com/zu/1346.html
- 转载请注明: : 萌小白 2023年1月15日 于 卖萌控的博客 发表
- 百度已收录
