随着测序成本的不断降低,测序通量的不断提升,基因组大数据时代已经到来。无论是拟南芥的 1001 基因组研究项目,还是 3000 株水稻基因组的公布,都让研究者们获得了大量可用的测序数据,同时,不同品系的动植物基因组也逐渐发布,如拟南芥的 col 和 Ler,水稻的 9311 和 Nipponbare,那么如何快速的比较两套不同品系的参考基因组,从而准确鉴别两者之间的差异 SNP & InDel 位点,以用于遗传标记的开发和品种鉴定呢?
在这里,小编向您推荐一套快速有效的解读思路,可以获得基因组之间大部分的差异位点。我们方案的入手点是重测序的基本原理,通过高通量测序获得的 paired-end reads 和参考基因组比对,检测其中纯合的 SNP 和 InDel,获得测序样本和参考基因组的差异位点。
但是我们现在只有两套参考基因组,该怎么进行呢?方法就是用参考基因组模拟测序数据,这里推荐一款经过 C++ 优化的测序数据模拟软件 -- ART
下载链接如下:
http://www.niehs.nih.gov/research/resources/software/biostatistics/art/
软件有多个系统版本,下载网页上和 readme 文件中也有详细的说明,以 linux 系统为例,按顺序执行以下步骤即可完成安装:
ART 的功能非常强大,可以模拟当今市面上主流的测序数据格式,此外,还可以设置测序深度,Insert-Size 的长度和标准差,测序质量值,是否含有 N 碱基等参数,几乎可以模拟您想要的任何测序数据。
当获得了模拟数据之后,就能往另一套参考基因组上比对了,这里就拿 9311 和 Nipponbare 举例。
1
利用 ART 软件模拟 9311 基因组 60X 的 illumina X-Tenpaired-end 测序数据,reads 长度为 150bp。Insert Size 为 400bp,标准差为 10,输出文件名为 9311.
生成的文件和数据量如下图
2
利用 bwa 软件 mem 模块将模拟数据比对到 Nipponbare 基因组,通过 samtools0.1.19 处理比对结果,如 samtools 版本太高,需要更改 samtools 的参数。

3
然后用 samtool smpileup 模块或者 GATK 检测 snp 位点。

4
通过 DP4 数值计算 SNP & InDel 的频率,由于是模拟数据,不会存在测序误差,所以 SNP 中频率为 1 的位点就是两套基因组的差异位点。
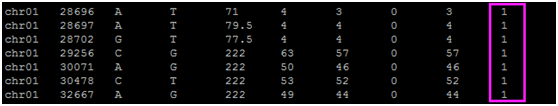
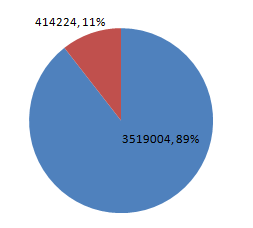
经过测试,这种方法检测出的SNP数量,是已经公布的 9311-Nipponbare 差异位点的 89%,且分布均匀。
- 本文固定链接: https://maimengkong.com/zu/1353.html
- 转载请注明: : 萌小白 2023年1月16日 于 卖萌控的博客 发表
- 百度已收录
