2020年, 扬州大学和 百迈客生物公司联合开发了基于ONT平台的植物转录组测序新技术,对ONT、PacBio和Illumina转录组测序进行了详细比较和综合评估。此次给大家带来数据的超精细解读~

摘要
在许多不同的研究领域中,使用Pacifc Biosciences(PacBio)和牛津纳米孔技术(ONT)进行第三代测序的研究数量正在迅速增加。与二代测序技术的短读长(50-500bp)相比,第三代测序的序列长度长(10kb以上),对解析复杂基因结构和重复序列等具有明显优势。ONT测序技术已在全基因组测序及表观遗传学领域大量应用,然而在全长转录组测序方面,使用较多的是Pacbio技术,ONT技术应用则较少,少量的研究主要集中在动物转录组测序上,而植物转录组ONT测序的应用与评价还没有报道。
为了评估ONT和Pacbio不同测序平台在植物转录组上的性能,我们选取植物模式物种拟南芥作为研究对象,对PacBio,Nanopore直接cDNA(ONT Dc)和Nanopore PCR cDNA(ONT Pc)测序的读数进行了详细评估,包括原始数据的特征和转录本的鉴定,同时与对应的Illumina数据进行比较。本文对拟南芥转录组的PacBio和Nanopore的RNA测序进行了全面比较,结果表明,ONT Pc在产生超长reads方面更具成本效益,可以体现转录本的特征并量化转录本的表达,因此,ONT Pc是一种用于植物全长转录组分析的经济高效的新方法。
结果
- ONT Pc 表现出更好的原始数据质量,而 PacBio 产生更长的读取长度
- 在转录组分析中, PacBio 和 ONT Pc 在转录本鉴定,简单的序列重复分析和长链非编码 RNA 预测方面表现相似。
- PacBio 在鉴定可变剪切方面表现优异,而 ONT Pc 可以估计转录本表达水平。
材料与方法
材料:拟南芥
测序平台:Illumina,PacBio,Nanopore
软件:SMRT-Analysis软件包v3.0,MinKNOW2.2中的Guppy,MISA,TransDecoder,CPC,CNCI,CPAT,Pfam,HISAT2和StringTie,Salmon
结果
为了比较RNA测序方法的性能,我们在Illumina NovaSeq,PacBio Sequel,Nanopore仪器上对拟南芥的cDNA文库进行了测序。此外,我们分别使用Nanopore GridION和Nanopore PromethION直接对cDNA(ONT Dc)和扩增后的cDNA(ONT Pc)进行了测序。
测序后,我们从每个Illumina RNA-Seq样品中获得了超过2100万次条clean reads,将每个样品的clean reads比对到参考基因组,总比对率>84.67%。
对于PacBio SMRT测序,构建了一个片段大小1-6 kb的全长cDNA文库,并在一个SMRT cell中进行了测序,最终获得了26.71Gb的clean date,516,364个高质量的ROI(插入片段)。这些ROI包括416,662(80.7%)个全长非嵌合(FLNC)reads和79,984(15.5%)个非全长(nFL)reads(图1),最后用ICE算法进行聚类,我们最终在拟南芥中获得了181,135个ccs一致性序列。
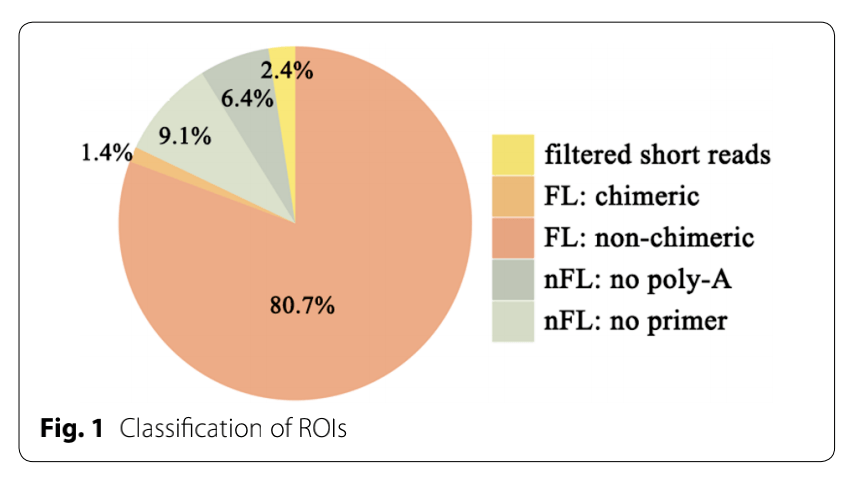
对于ONT Dc测序,我们分别从CTRL1,CTRL2和CTRL3获得了6,892,169、5,687,972和10,936,056条clean reads。这些clean reads的N50值分别为1245、1438和1345bp,平均长度分别为1065、1228和1167 bp。我们将两端有引物的序列鉴定为全长序列,我们分别从CTRL1,CTRL2和CTRL3获得了128,781、138,295和262,832个全长序列。
对于ONT Pc测序,分别从CTRL1,CTRL2和CTRL3获得了8,146,264、7,713,840和6,912,956条clean reads。这些clean reads的N50值分别为1252、1292和1270bp,平均长度分别为1222、1246和1225bp,其中,分别从CTRL1,CTRL2和CTRL3获得了5,682,227、5,563,209和5,207,164条全长序列。
一、PacBio 和ONT结果的原始数据比较
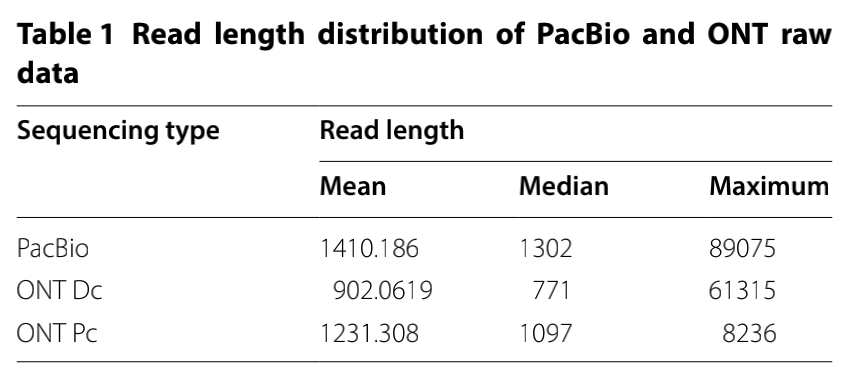
为了比较来自PacBio和ONT测序的原始数据,我们从PacBio中随机选取了10 Mb原始reads(3,112,439 subreads),并从每个ONT样本中选择了100,000个1D reads(总计300,000个)。PacBio reads的平均长度为1410.186 bp,中值和最大读长分别为1302 bp和89,075 bp。ONT Dc数据较短,中位长度和最大长度分别为771 bp和61,315 bp。ONT Pc数据的中值和最大长度分别为1097 bp和8236 bp(表1),PacBio和ONT表现出显著差异(图2a–c)。与PacBio相比,ONT Dc数据的长度分布偏向左侧,其中很大一部分读数小于2000 bp(图2a–c),ONT Pc数据的长度分布类似于ONT Dc数据的长度分布(图2b,c)。
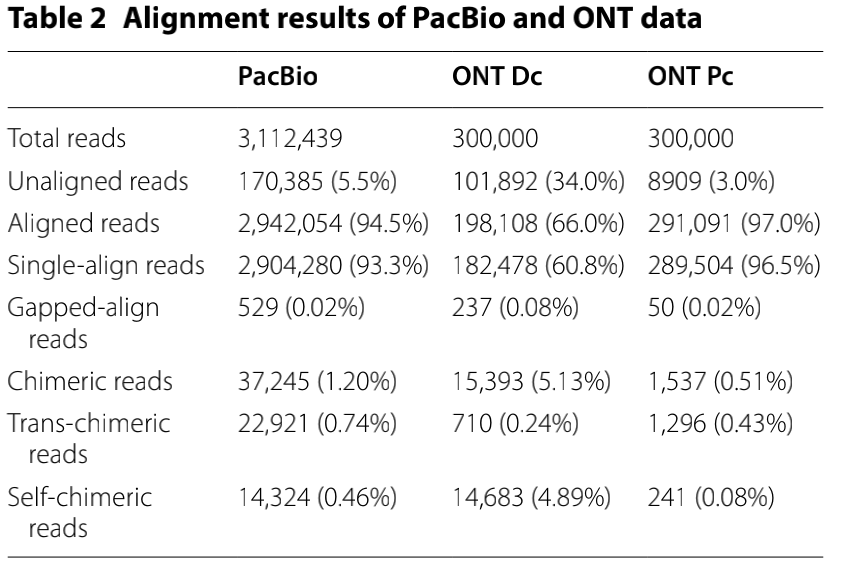
长读长序列的比对率对于确认重复元件、基因亚型和基因融合至关重要。在PacBio的subreads中,有94.5%与参考基因组比对上(图2d,表2),与PacBio的subreads相比,ONT Dc 1D reads的比对率较低(66%)(图2e,表2),ONT Pc 1D reads的比对率较高(97%)(图2f,表2)。对于PacBio和ONT Pc数据,我们发现读长较短的序列(<500 bp)具有较低的比对率(图2d,f),这可能是此类短读数据中的接头和连接序列占了较大部分。但是,在ONT Dc 1D读取中,所有长度的比对率均相似(约60%)(图2e)。
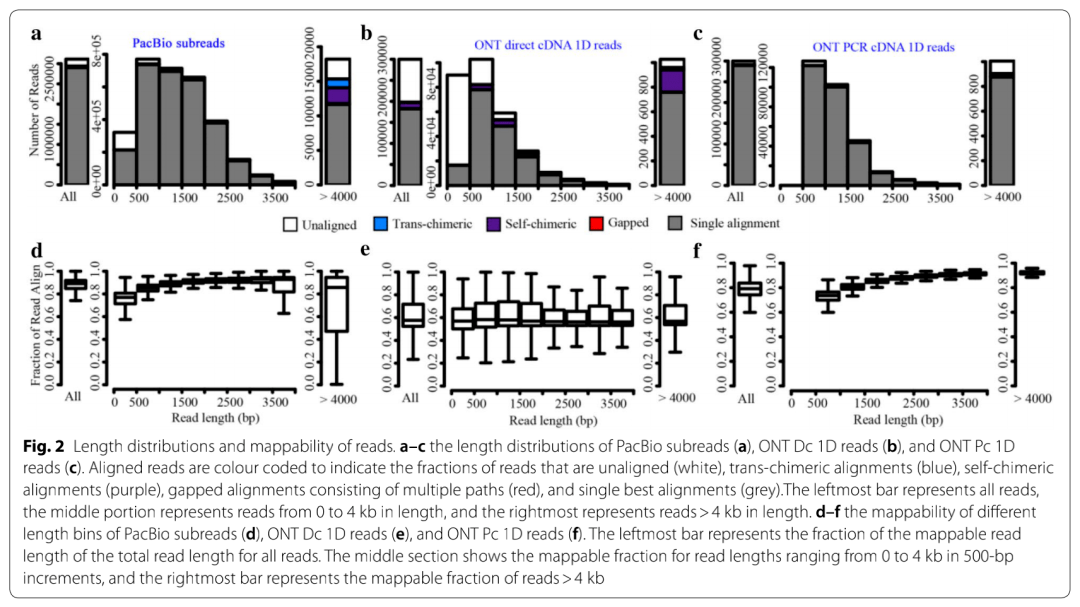
长读长的某些区域可能特别容易出现错误,并且长读取可能比对到分离的片段,称为"gap-aligned reads"。基因融合或反式剪接产生的长读长可以比对到分离的基因组位点;这些被称为“trans-chimeric reads”(反式嵌合序列)。PacBio subreads包含0.74%的“trans-chimeric reads”,而ONT 1D数据包含的更少一些(ONT Dc:0.24%,ONT Pc:0.43%)(表2)。此外,PacBio subreads在长读长片段(>4 kb)中显示出更高的反式嵌合率(图2a),由于未能从原始数据中删除接头序列,两端长读长片段可能会被比对到相同的基因组位点,称之为“自嵌合序列”。PacBio subreads 和ONT Pc 1D reads分别包含0.46%和0.08%的自嵌合序列,出乎意料的是,ONT Dc 1D reads 比例更高(4.89%)(表2),嵌合序列可能会过高的估计DNA分子的长度。
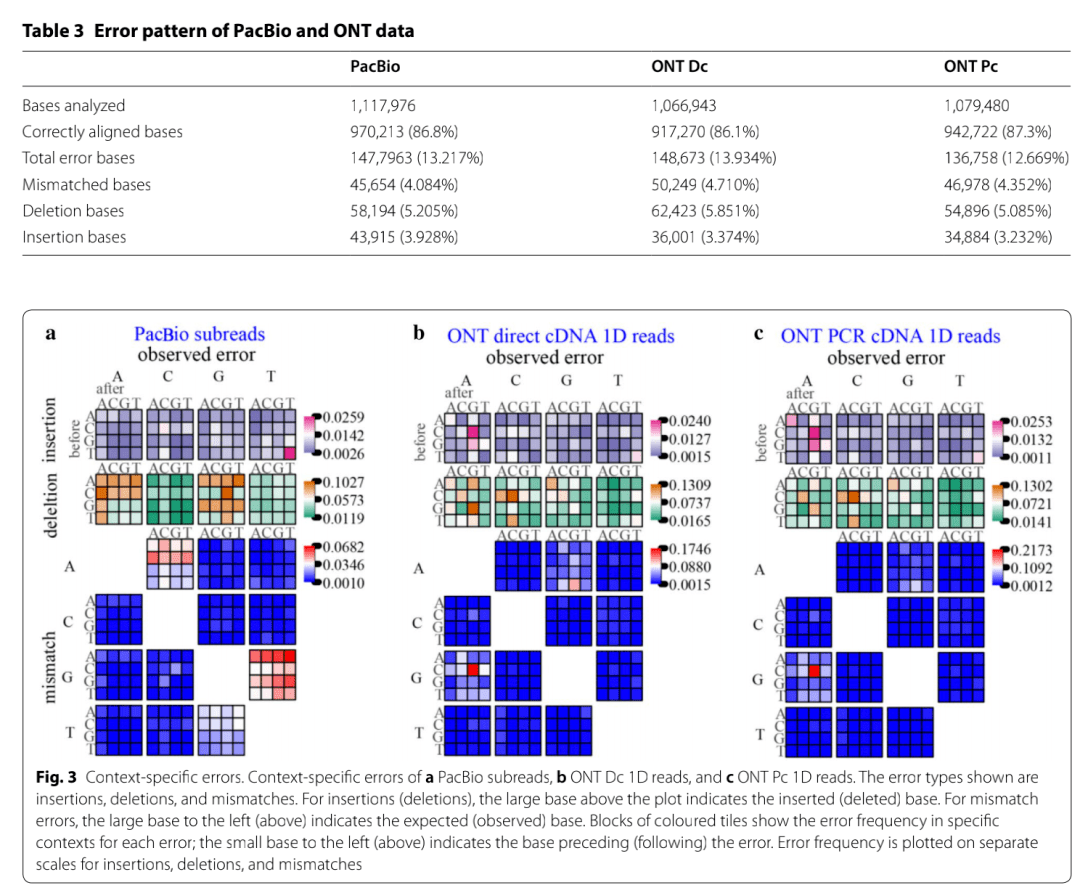
错误率和错误模式可以体现数据的质量,这对单核苷酸分辨率分析有很大的影响。PacBio的错误率为13.217%。与PacBio相比,ONT Dc数据的错误率略高,达到13.934%,而ONT Pc数据的错误率较低(12.669%)(表3),这表明ONT Pc数据的基本质量比PacBio数据略高。
此外,PacBio和ONT错误的组成相似。对于PacBio,ONT Dc和ONT Pc数据,错配的比例分别为4.084%,4.710%和4.352%(表3)。在PacBio,ONT Dc和ONT Pc数据中,缺失率都比较高,分别为5.205%,5.851%和5.085%,插入率为3.928%,3.374%和3.232%。
综合考虑,插入和缺失(indels)是导致PacBio和ONT数据错误的最大原因,PacBio和ONT的数据错误都表现出特定的模式,在PacBio序列中,大多数错配是由几个特定的模式导致的,例如GA→TA,GC→TC,GG→TG和GT→TT(图3a)。CG→CA的不匹配在ONT Dc数据中最丰富,其次是AG→GG(图3b),ONT Pc数据中CG→CA也是最丰富的(图3c)。此外,在PacBio序列中最常见的是插入的T和缺失的A和G(图3a),而插入的A和删除的A和C在ONT数据中最常见(图3b,c)。
二、Illumina ,PacBio,和ONT转录本数据
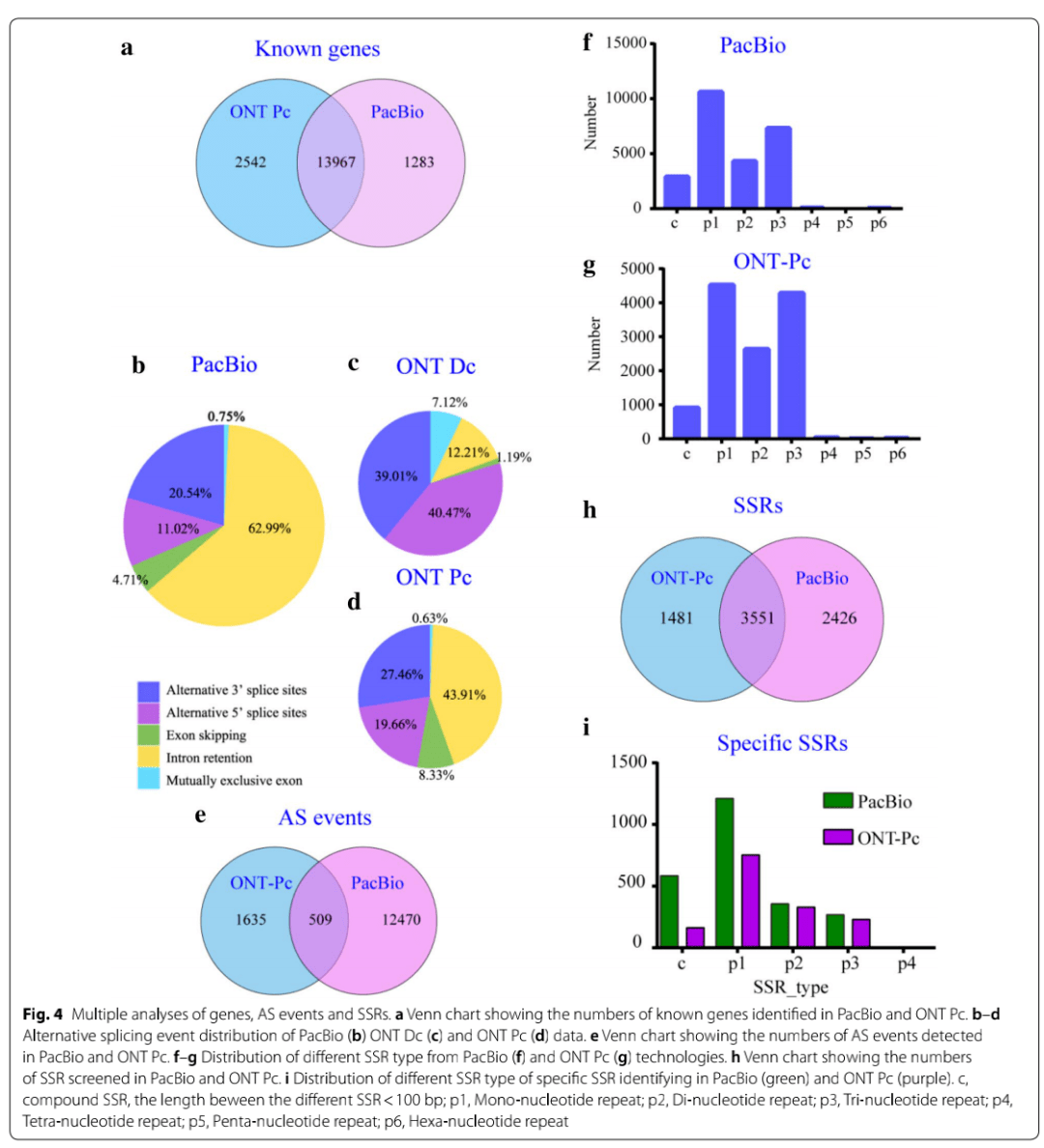
基于Illumina的短读长reads,获得了总共21,157个基因。对于PacBio测序,使用非全长序列对转录本进行了一致性校正,并获得了129,080个高质量的转录本。使用GMAP软件将经过校正的转录本定位到参考基因组,去除冗余序列后,从13,376个基因位点生成了38,011个非冗余转录本。对于ONT Dc测序,在校正所有全长序列后,将校正后的转录本比对到参考基因组上,从而共产生了47,601个转录本,同样,ONT Pc测序共有36,775个非冗余的转录本比对到参考基因组上。
三、AS (可变剪接)检测
在PacBio的unique mapped reads中,我们检测到总共12,979个可变剪切,包括97个互斥的外显子,8175个内含子保留(IR),611个外显子跳跃(ES),1430个5'端可变剪切(Alt 5')和2666个3'端可变剪切(Alt 3')。在PacBio数据中,最常见的可变剪切是IR(62.99%),其次是Alt 3′(20.54%),Alt 5'(11.02%),和ES(4.71%);几乎没有发现相互排斥的外显子事件(0.75%)(图4b)。
在ONT Dc数据中检测到的AS事件就少得多了,CTRL1,CTRL2和CTRL3分别包含1433、928和4367个可变剪切,每种可变剪切类型占比也与PacBio数据不同。ONT数据中最容易检测的可变剪切是Alt 3',其次是Alt 5',IR和互斥的外显子;ES最少(1.07%)(图4c)。
相反,在ONT Pc数据中,分别在CTRL1,CTRL2和CTRL3中标识了1897、2048和2034 个AS事件,每种可变剪切类型占比均与PacBio数据的分数相似(图4d),PacBio和ONT Pc中只有509个常见的可变剪切(图4e),包括170个Alt 3'(33.40%),62个Alt 5'(12.18%),44个ES(8.64%)和233个IR(45.78%),结果表明,ONT Pc在可变剪切检测方面能力相对较弱。
四、SSR 检测
我们选择长度>500 bp的转录本共58,885个序列(122,942,629 bp)使用MISA进行了SSR分析。结果,从PacBio数据中鉴定出共29,394个SSR和20,243个复合SSR序列。含有1个以上SSR的序列有6067个,复合形成的SSR有4109个。此外,SSR位点重复单元为1~6个碱基,SSR基因座的重复单元为1〜6个碱基,其中单核苷酸重复序列最多(p1:10623,42.01%),其次是三核苷酸重复序列(p3:7316,28.93%)和二核苷酸重复序列(p2:25285,17.10%),4个碱基和更多的重复单元相对较少(图4f)。
五、 新转录本的CDSs 和lncRNA预测
使用TransDecoder(v3.0.0),在PacBio数据中鉴定出31,137个ORF,其中25,256个是完整的ORF。此外,在ONT Dc和ONT Pc数据中分别预测到33,419和15,968个完整的ORF,图5a展示了完整ORF的CDS长度分布。在PacBio数据中,完整ORF的CDS长度大部分在100到1000 bp之间(图5a);在ONT Dc和ONT Pc数据中,长度分布向左倾斜。在ONT Dc数据中,完整ORF的大多数CDS长度为0–100 bp,其次为100–200 bp,只有少数>200 bp;在ONT Pc数据中,完整ORF的CDS长度范围为0到800 bp,大多数为0-300 bp(图5a)。
使用CPC,CNCI,Pfam和CPAT四种方法分别在PacBio,ONT Dc和ONT Pc数据中分别预测了257、8911和249个lncRNA(图5b-d),在PacBio和ONT Pc数据中都鉴定出了35种常见的lncRNA。我们随机选择了16种独特的lncRNA(PacBio数据中的8个lncRNA和ONT Pc数据中的8个lncRNA)进行PCR扩增和Sanger测序验证,来自PacBio数据的8个lncRNA中,有2个与RNA-Seq序列完全相同,有2个具有少于3个错配的核苷酸(图6a,b);从ONT Pc数据中选择的8个lncRNA中,有5个lncRNA被验证,所有的lncRNA错配核苷酸都少于3个(图6a,c)。
六、通过ONT 和Illumina数据估算异构体丰度
我们评估了ONT和Illumina数据在转录本定量中的作用,使用FPKM和CPM值分别量化Illumina、ONT Dc和ONT Pc数据的转录本表达水平,进一步计算了每个重复的Illumina数据和ONT Dc数据之间的相关性。结果表明,CTRL1,CTRL2和CTRL3 Illumina和ONT Dc数据之间的表达相关性分别为0.747、0.719和0.711(图7a–c);Illumina和ONT Pc之间的表达相关性较高,CTRL1,CTRL2和CTRL3分别为0.932、0.928和0.923(图7d-f)。
结论
文:ZS
排版:市场部
转自:百迈克
- 本文固定链接: https://maimengkong.com/zu/1354.html
- 转载请注明: : 萌小白 2023年1月16日 于 卖萌控的博客 发表
- 百度已收录
