撰写: 小高 来源:小张聊科研平台的“ i生信”公众号,微信公众号搜索“ i生信”即可关注/扫描关注见文末
#生信分析# #生信发文#
今天笔者带来的是一篇整合分析RNA-Seq和ChIP-Seq的文章。RNA-seq是用于分析不同细胞类型和状态的差异基因表达的重要工具,基因表达受到多种机制的调控,包括表观遗传学上的翻译后的组蛋白修饰,这可以通过ChIP-Seq获得。单独分析RNA-Seq或ChIP-Seq不能完全解释复杂的潜在的基因表达调控机制。定量整合RNA-Seq和不同条件下的组蛋白修饰的ChIP-Seq数据能够提升对基因调控中表观遗传的理解。理论上来讲,整合分析RNA-Seq和ChIP-Seq是研究基因调控的唯一选择。然而,在技术上仍然还处于早期。作者引入intePareto用于整合分析RNA-Seq和ChIP-Seq数据。IntePareto能够实现在基因水平上整合RNA-Seq和ChIP-Seq数据集,能够定量RNA-Seq和ChIP-Seq,得到组蛋白修饰与基因表达的相关性,并且能够基于RNA-Seq和ChIP-Seq改变的一致性来寻找优先候选的基因。IntePareto包括匹配,整合和优化三个部分(Fig 1a)。
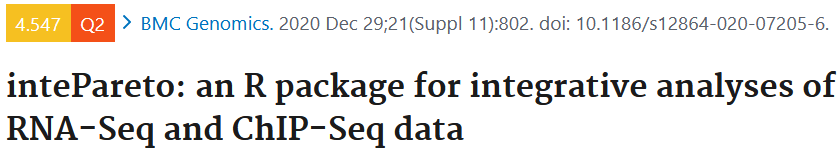
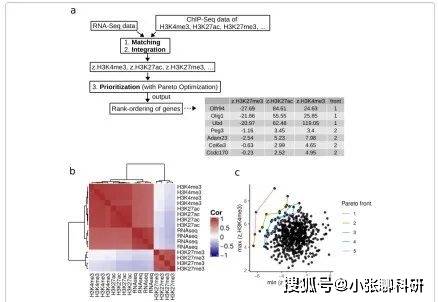
结果一:利用公开的RNA-Seq和ChIP-Seq数据评估intePareto
作者利用一个已经发表的Tet2敲除鼠与野生鼠的胚胎干细胞的研究中的RNA-Seq和ChIP-Seq数据来评估intePareto。Tet2被认为参与增强子区的DNA甲基化调控。作者期望能够从Tet2敲除和野生的胚胎干细胞中找到表观组和转录组一致的改变。针对每个细胞类型,数据包括RNA-Seq data 和 ChIP-Seq(包括了H3K4me1, H3K4me3, H3K9me3, H3K27ac, H3K27me3和 H3K36me3)生物学重复。
结果二:数据处理
原始RNA-Seq数据的FASTQ文件与ENSEMBL中的参考转录组比对,之后利用Kallisto进行表达定量。原始ChIP-Seq数据的FASTQ文件利用BWA (0.7.17)与ENSEMBL中的参考基因组比对,结果文件利用Samtools (version 0.1.19)进行排序和相应的index文件构建。
结果三:intePareto分析
已知组蛋白marker H3K4me3, H3K27me3和 H3K9me3富集在基因的启动子区,其他的marker,比如H3K4me1和 H3K27ac,经常与基因增强子和活化的启动子区相关,而H3K36me3与gene body区相关。为了定义与基因启动子相关的表观信号marker,作者计算ChIP-Seq数据在特定基因的启动子区的reads数;针对H3K36me3这个marker, 作者计算在gene body区的read数。整合RNA-Seq和ChIP-Seq数据采用highest策略(doIntegration function)。作者得到有意义的表观遗传和转录组信号,基因表达与激活marker正相关,与一直maker负相关(Fig 2)。之后进行优化,活化的组蛋白markers(H3K4me1, H3K4me3, H3K27ac, H3K36me3) 对应高的正Z-score值;抑制的组蛋白markers(H3K9me3, H3K27me3)对应低的负Z-score值。最终结果的gene列表根据
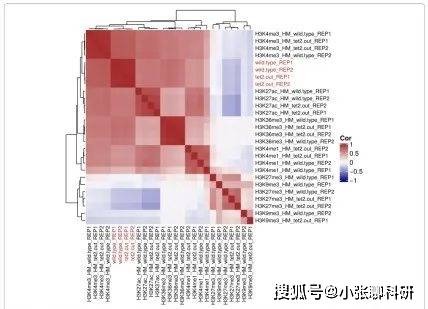
结果四:intePareto结果的下游分析
IntePareto结果中排在前面的基因进行GO富集分析,富集的结果都富集到已知的Tet2的功能特征。尤其是,Tet2可以通过改变DNA甲基化状态来影响胚胎干细胞分化和增殖,特别在神经分化,心脏和其他器官的分化过程中。Fig3b展示了前14个基因的热图。上调和下调基因展现了不同的模式。图中可以看出H3K27me3和H3K9Me3有相似的功能,H3K4me1, H3K4me3, H3K27ac和H3K36me3有相似的功能,这与先前报导的结果一致。值得一提的是Eif2s3y基因,它在Tet2敲除胚胎干细胞中高度下调,但是,当只利用RNA-Seq数据分析的时候,它并不是显著下调的。然而,在我们整合分析中,Eif2s3y基因排在前二。这也表明整合分析的优点,能够降低只利用一种数据的假阴性和假阳性率。
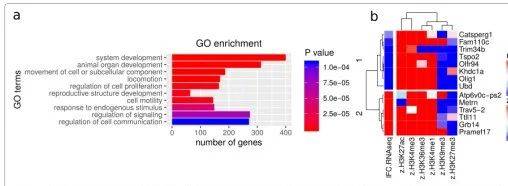
结果四:与已经存在的方法比较
为了评估intePareto的性能,作者与一个最近发表的一个基于层次贝叶斯模型的聚类方法进行比较,还与单独的RNA-Seq结果比较。比较的质量指标,作者选用的是感兴趣的GO term的富集得分。感兴趣的GO term是指之前研究确认的,比如神经发生,心室发育,乳腺形成和肢体形态发生。IntePareto方法和基于模型的方法可以富集到神经发生(Fig 4a)和肢体形态发生(Fig 4d)。仅RNA-Seq的结果不能富集到这些功能。IntePareto方法能够富集到心室发育(Fig 4b)和乳腺形成(Fig 4c),这些功能不能通过单独的RNA-Seq或者基于模型的方法得到。GO富集的替代方法,pathway富集,可以产生补充信息。

笔者总结
多组学的整合分析已经是一个必然的趋势,如何实现不同组学数据直接的融合是一个亟待解决的问题。这篇文章结合转录组以及表观遗传组用于研究基因表达调控。当然基因的表达调控不仅仅是这两个组学的结果,还包括染色质重塑,转录因子,DNA甲基化,甚至RNA修饰等的作用。因此,融合多组学研究的方法需要进一步的发展,用于解析复杂的生物学事件。
原文链接:https://mp.weixin.qq.com/s/zKMatnwpa0B5XU8gzH2GzA- 本文固定链接: https://maimengkong.com/image/1350.html
- 转载请注明: : 萌小白 2023年1月15日 于 卖萌控的博客 发表
- 百度已收录
