做个铺垫
上次写的表观遗传学第一弹“如何研究DNA甲基化”的推文,反馈不错,小编还是有点小欣喜的!
今天,小编打算把上篇提到的DNA甲基化的BS技术单独提出来,跟大伙儿详细介绍一下基于该技术的数据分析流程及实现。
读过上篇的童鞋们已经了解到,BS技术就是基于亚硫酸盐将胞嘧啶C转化成为胸腺嘧啶T并测序。
上篇推文入口 >>
Lister等在2008年首次通过该技术研究拟南芥和人的全基因组甲基化,拉开了亚硫酸盐 DNA甲基化测序的序幕!
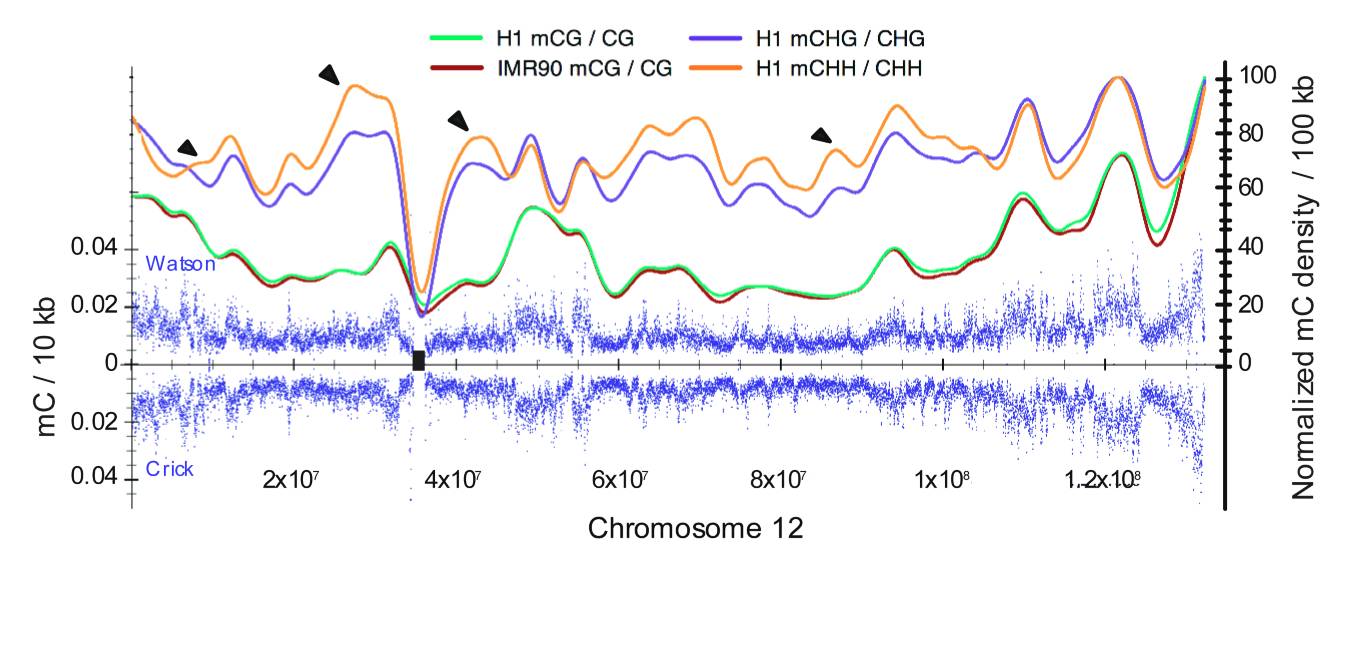
12号染色体上甲基化率的分布情况
然而,测序的花费,即便是放在现在来看,也不便宜。比如,人的基因组是3G,按照饱和测序,每个样品30X计算,基本上每个样品都需要一个lane的数据(约90G)。如果做大规模样品测序,总成本也是很高的,并非每个实验室都能承受。
为了解决这个问题,基于亚硫酸盐转换的各种新技术也相继问世。简化基因组甲基化测序(RRBS)就是其中的一种。基因组首先通过MspI酶切。在人基因组中,其识别位点CCGG大概有3M,实际检测到的位点约1.5M~2M之间。
RRBS的建立是基于SE36测序,确保每个测序片段都会检测到1个CG位点,因此其选择的DNA片段无需太长,通常为40-220bp。
然而,新的问题又来了!随着测序技术的发展,从原来的PE50,到highseq2000的PE75、PE100,再到现在的X TEN的PE150,如果酶切的片段短,很容易就会测通,导致大量的数据浪费,而检测的位点数却并没有增加。随着测序读长逐步增加,数据浪费的问题也就越发严重,这也是现在很多测序服务公司不推广RRBS的原因。
另外呢,在大规模样品筛查的时候,RRBS可能覆盖不到想要的基因区域,导致很多时候都筛选不到相应的基因biomarker。其实同样的问题,也存在于450k甲基化芯片和850k甲基化芯片中。
包括小编发表的基于双酶切DRRBS技术,虽然可以通过变换不同的酶切组合,同时改变插入片段的长度范围,增加基因组的覆盖和CG位点数目(>10M CpG位点,是常规技术覆盖CpG位点数的5倍左右),但仍然不能完全解决调控区域(启动子,CGI,CGS,enhancer和转录因子区域)覆盖不全的问题。
这么看来,全基因组甲基化(WGBS)太贵,RRBS/DRRBS又可能覆盖不到感兴趣的基因调控位点(当然,RRBS技术作为简化基因组测序的研究策略也有其独特的优点,在这里不展开讲,小编也有开发相应的oxRRBS技术,感兴趣的话,后面可以做一个专题专门介绍一下),敢问,对于经费没那么充足的实验室来说,到底路在何方?
说起来有点小自豪,小编所在的团队首次提出了将液相杂交捕获和亚硫酸盐测序相结合的理论,可以使DNA甲基化研究更加灵活,成本更低,多样本分析的时候也能保证覆盖的均一性和稳定性。各位可以参考小编发表的技术方法,直接合成相应的探针,就能在自己实验室研究目标基因的甲基化水平了。
Tips
给公众号回复“启动子技术”,可直接获得相关论文的全文。
不过,如果你的研究目的是筛查甲基化biomarker,小编仍然建议你先把全基因组的调控区域筛选一遍,推荐使用LHC-BS(启动子液相捕获甲基化芯片)技术。该技术的目标区域呢,除了普遍关心的启动子(所有注释基因TSS上2Kb,下游500bp),还覆盖了CpG岛和CpG shore等多种调控元件。跟850K芯片相比,费用相差不多,但是检测的位点多出2倍,结果也更准确(话说,50X~70X的数据能不准确么)。
唠叨了这么多,切入正题,我们还是以液相捕获甲基化测序(LHC-BS)为例,一起来聊聊BS技术的常规分析方法。
BS技术常规分析方法
关于过滤及比对
所有BS类数据的实验思路都是类似的,即对DNA片段进行中亚硫酸盐处理,然后对转化后的片段进行上机测序,针对得到的测序reads,进行质量控制。一般来说,我们称下机数据为原始数据(raw data),过滤后的数据为clean data。
在过滤的过程中,通常会针对低质量的测序reads过滤,过滤规则一般是过滤掉Q20base数小于某个阈值(如50%)的reads,或者测不准的碱基个数>50%的reads。不过这一类的reads非常少,一般只能占总reads数的1-5%。
然而,在包lane测序的时候,因为经过bisulfite处理,DNA片段的复杂度会降低,总体测序质量会下降,需要做实验方面的优化,或者与正常基因文库混合上机测序,效果会好一些。在这个部分,可以自己写脚本处理,也可以用写好的软件,如华大开发的SOAPnuke等软件处理。
去掉测序质量低的reads后,下一步就来到最消耗计算资源的步骤了:比对。
以前,没有专门的软件工具可用,这个部分的处理非常费事。最初大家是用SOAP,或者BWA比对软件来比对,操作过程是将参考序列进行一次C转化为T,比对一次,然后再将参考序列的G转化成A,再比对一次,操作很是麻烦。
现在可直接使用的比对软件已经很多了,比如BSMAP、bismark等软件,操作上很是简洁。就性能而言,个人还是推荐BSMAP,使用方法举例如下:
./bsmap -a 3.clean.1.fq -b 3.clean.2.fq -m 0 -x 900 -d hg38.lambda.spike.fa -o 3.sam -n 0 -v 0.1 2>3.log
用法上也非常简单,自己查询一下帮助文档就能上手。比对之后,可以得到sam文件,并通过samtools转化为bam格式。Samtools还有sort排序、去冗余、统计基本比对信息等功能,感兴趣的话,可以在网上自己找找教程,这里就不赘述了。
Tips
早先,项目做完发表文章的时候,需要上传比对的文件,现在已经不用了,所以可直接删除sam,中间过程的bam文件,节省存储空间。
关于测序饱和度
关于测序饱和度的问题,WGBS技术可以预计30X即可。现在的比对率一般为90%以上,差不多每个位点上的胞嘧啶有效覆盖大概也有10X以上了。
Tips
正负链各自带有甲基化信息,所以参考序列中的C和G(原始负链C)都带有甲基化信息,正负链上每个胞嘧啶的甲基化信息深度为总覆盖深度的一半。
其他技术,如RRBS,可通过酶切位点的覆盖度来评估,而LHC启动子甲基化捕获技术,可以通过探针的覆盖度来评估。
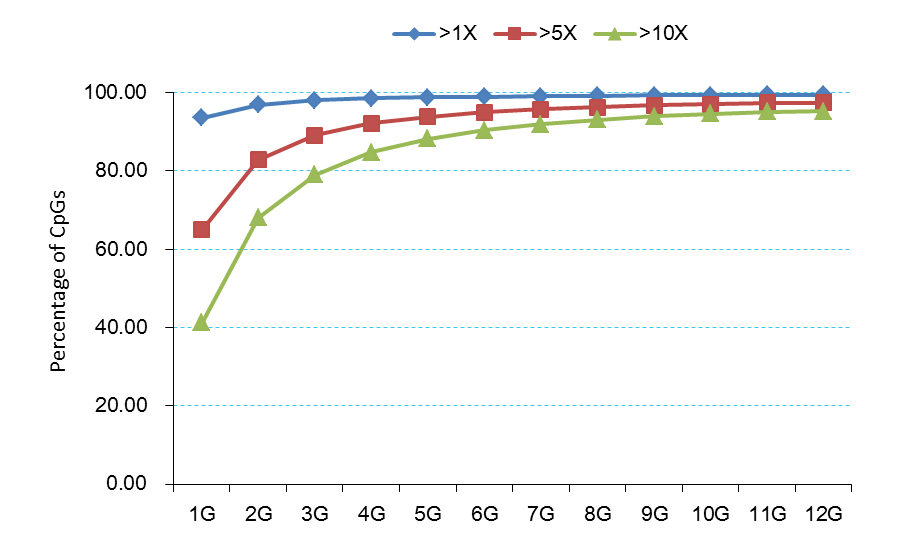
LHC测序量和胞嘧啶覆盖深度变化曲线
关于甲基化率及注释问题
接下来我们聊聊如何得到每个位点的甲基化率信息。
BSMAP软件有一个python的脚本,非常简单好用(这也是很多公司说自己可以处理甲基化数据的原因),这里给出来一个例子,具体参数大伙儿可以自己研究一下:
methratio.py -d hg38.lambda.fa -c Control -o 9.Control.meth.txt -u -p -z -q -m 1 -s samtools-0.1.19/ -i correct 2.bam
得到胞嘧啶位点的甲基化率之后,就可以算DMR了。可用的相关软件也非常多,常见的有基于泊松检验的算法,以及QDMR(http://bioinfo.hrbmu.edu.cn/qdmr)、eDMR (https://code.google.com/p/edmr/)等。
这里小编重点推荐metilene软件
(参考文献:)。
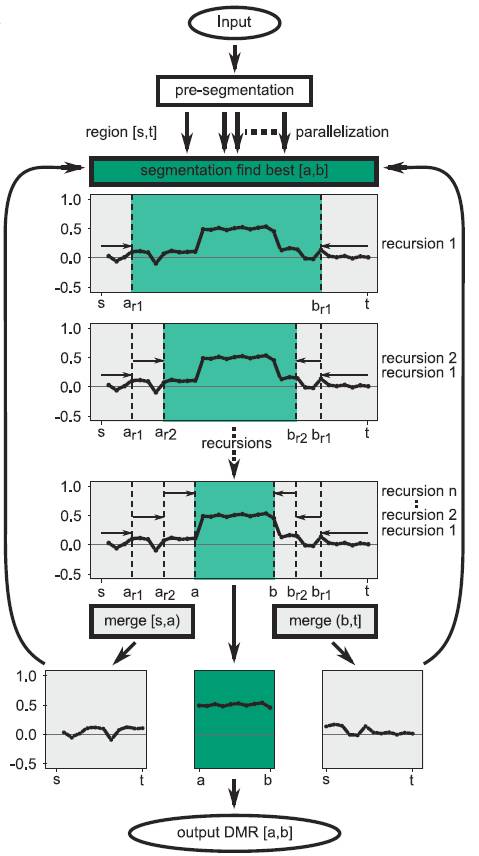
这个分析软件的模型挺有意思的。由于软件不基于分布模型和背景模型,可以在没有进一步参数修正调整的情况下适用于WGBS和RRBS。这种方法基于一种循环的二进制片段(CBS) (Siegmund 1986; Olshen et al. 2004),扫描成对的平均甲基化率差异信号的改变位点,也就是说,通过CpG平均甲基化率的水平组间差异,来界定同类甲基化差异区域。
随后,间隔利用2维的Kolmogorov–Smirnov test (2D-KS test,Fasano and Franceschini 1987)检验,先将基因组分割成片段,避免没有甲基化信息的长的延伸片段被包含进来call DMR这些区域,按照片段递归,直到区间包含少于用户定义的最少CpG个数(一般是5个),或者直到没有p值改善的情况。
具体模型如下图所示。
得到DMR之后,下一步就是注释到相应的基因上,并进行GO分析、KEGG分析,以及PPI(蛋白相互作用网络分析)分析,这方面的资料也非常多,就不用小编再啰嗦了。
关于参考基因组校正
接下来,我们聊一个很多人关心的问题:在很多样品中,个体的基因组和参考基因组是有差别的,这个特异性在甲基化数据分析中如何处理?
这个问题在很多公司提供的报告中并没有提及。事实上,BS数据在处理过程中也可以得到突变的数据。
甲基化测序过程中,因为原始序列的正负两条链的甲基化信息不同,所以在之后打断及PCR扩增等过程中,原始序列正负链是可以分开的。于是,我们可以通过正链的信息去call SNP,从而得到准确的ATG的甲基化信息。通过负链可以得到ATC的信息(Note:这里最好要分开两条链分别call SNP),这样就可以通过SNP的矫正来修正参考基因组了甲基化信息了。
项目设计中的一些问题
最后,小编想跟大伙儿说一下项目设计中的一些小问题。
很多项目设计都是case/control组。在得到每个位点的甲基化信息后,分析组间差异之前,首先需要先通过聚类查看一下样品之间的甲基化pattern的距离关系。特别是对于组间分析而言,需要首先确定组间的距离是否大于对照样品之间的组内距离。在满足组间距离更大的情况下才能进行组间DMR的寻找。
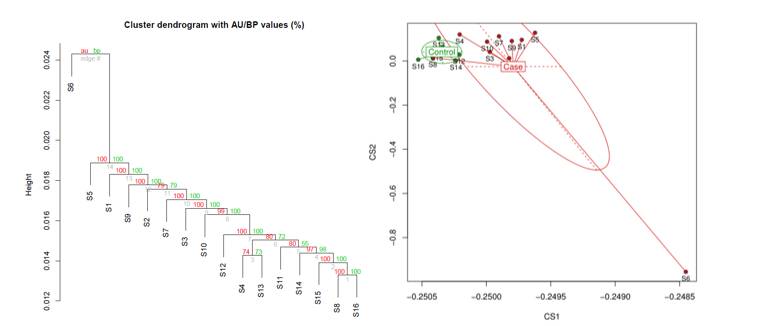
样品间启动子区域甲基化率聚类情况(左边是基于bootstrap算法的聚类情况,右边是基于PCA的展示)
上面的图呢,来自于真实的分析项目,是研究儿童受到某种重金属污染后的DNA甲基化的变化情况。
如图所示,上图中1-10号样品是case样品,11-16为对照样品。可以看到正常样品基本可以聚类到一起,而case样品也可以聚类到一起的,这说明启动子区域的甲基化在组间的差异是大于组内差异的。
在信息处理过程中,要注意的是,记得把已知的差异因素排除掉(如性染色体等),否则结果就会不够准确哦。就聚类关系而言,启动子甲基化技术的优点在于我们选择的区域是调控区域,DNA甲基化的修饰会更明显一些。
小结
其实BS类的基础分析流程还是很简单的,有现成的软件,重点在于在整体项目的把控方面。一定要记得先通过聚类的算法,查看组间和组内的关系,再考虑两组之间如何分析。如果组间距离大于组内距离,可以考虑将case和control组合并到一起,如果不是,最好是按照单个样品来比较分析。这一点在很多公司提供的结果中是不会考虑的,但是作为一线的科研人员,还是要考虑一下的。对于项目设计的更多细节,需要详细了解的小伙伴,可以联系小编咨询。
今天就说到这里,下次我们继续聊聊IP(MeDIP,CHIP-SEQ,RIP,MeRIP等)类技术的分析过程中遇到的问题,不见不散哈。
- 本文固定链接: https://maimengkong.com/zu/1351.html
- 转载请注明: : 萌小白 2023年1月15日 于 卖萌控的博客 发表
- 百度已收录
