2023
06-17
06-17
【生物信息学笔记】欧洲生物信息中心(EBI)资源
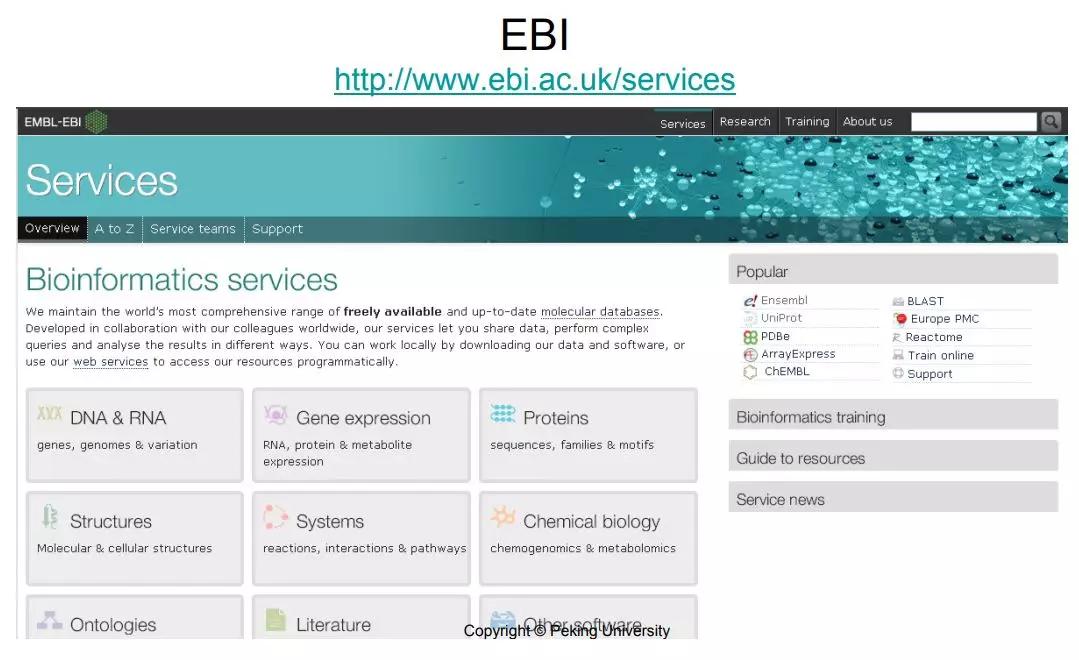 我们接着讲生物信息学的资源,之前我们介绍了NCBI,欧洲也有一个非常大的centralized resource,叫EBI 。和NCBI有点类似,也是一个包含各种各样数据库这样的资源,它主要也是针对从序列到蛋白到蛋白结构到表达到通路到Ontology各个方面。首先在DNA,RNA和蛋白的方面,基因和基因组最主要是有一个ensembl,很多人不知道EBI也会知道ensemb...阅读全文>&g... 阅 读 全 部 >
我们接着讲生物信息学的资源,之前我们介绍了NCBI,欧洲也有一个非常大的centralized resource,叫EBI 。和NCBI有点类似,也是一个包含各种各样数据库这样的资源,它主要也是针对从序列到蛋白到蛋白结构到表达到通路到Ontology各个方面。首先在DNA,RNA和蛋白的方面,基因和基因组最主要是有一个ensembl,很多人不知道EBI也会知道ensemb...阅读全文>&g... 阅 读 全 部 >
 2022年10月4日, Nature Genetics在线发表了德国莱布尼茨植物遗传与作物研究所(IPK)Jochen C. Reif和Martin Mascher联合团队及其合作者题为“ Genomics-informed prebreeding unlocks the diversity in genebanks for wheat improvement ”的研究论文。该研究通过对大量......
2022年10月4日, Nature Genetics在线发表了德国莱布尼茨植物遗传与作物研究所(IPK)Jochen C. Reif和Martin Mascher联合团队及其合作者题为“ Genomics-informed prebreeding unlocks the diversity in genebanks for wheat improvement ”的研究论文。该研究通过对大量......  AI & 癌症研究多组学技术和人工智能算法的同步发展推动了癌症精确医学的发展。2023年1月《 Seminars in Cancer Biology 》发表了一篇综述文章, 全面总结了基于人工智能的多组学肿瘤分析的最新进展,重点介绍了基于人工智能的多组学技术在癌症诊断、分类、早期筛查、反应评估和预后预测方面的应用。多组学技术随着高通量生物技术的发展,已经开发了多种组学技术来表征不...阅读...
AI & 癌症研究多组学技术和人工智能算法的同步发展推动了癌症精确医学的发展。2023年1月《 Seminars in Cancer Biology 》发表了一篇综述文章, 全面总结了基于人工智能的多组学肿瘤分析的最新进展,重点介绍了基于人工智能的多组学技术在癌症诊断、分类、早期筛查、反应评估和预后预测方面的应用。多组学技术随着高通量生物技术的发展,已经开发了多种组学技术来表征不...阅读... 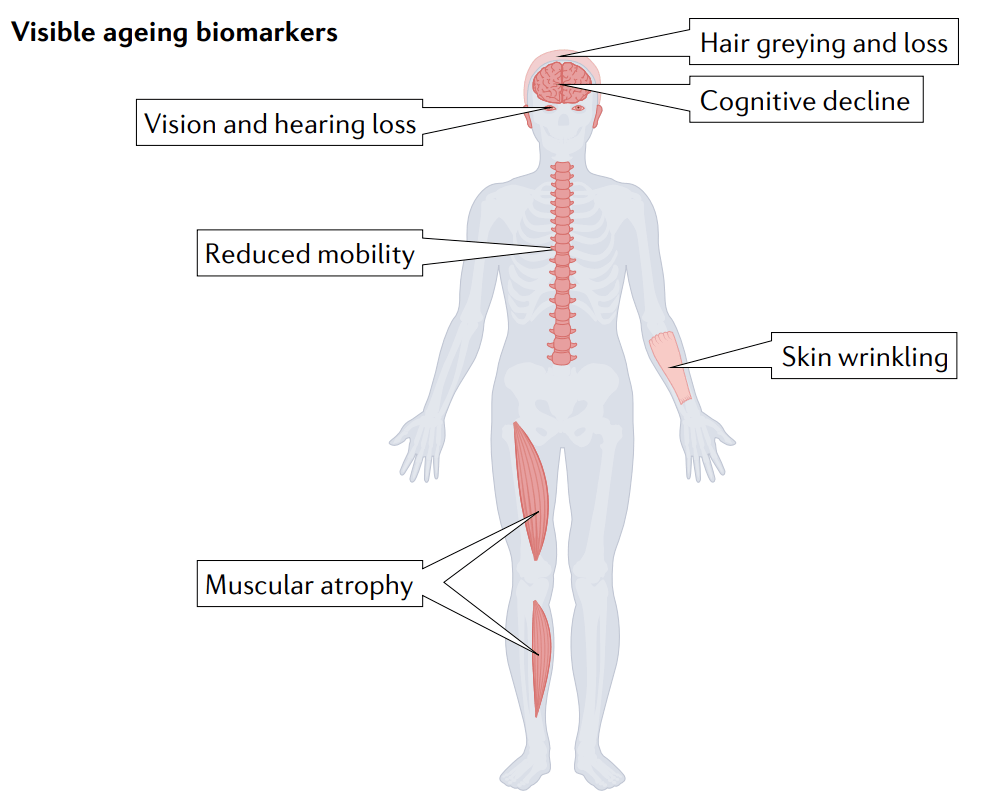 机器学习的兴起为大数据时代掀起新的浪潮。机器学习是人工智能的一个子集,通过泛型算法从数据集中发现模式和相关性并建立逻辑,并根据数据分析结果做出最佳决策和预测。目前机器学习已大量应用于各行各业,助力各产业或科学研究的深度提升。美国加州斯坦福大学的Tony Wyss-Coray团队于2022年6月在Nature Reviews Genetics上发表的一篇名为“Measuring biolo...阅读...
机器学习的兴起为大数据时代掀起新的浪潮。机器学习是人工智能的一个子集,通过泛型算法从数据集中发现模式和相关性并建立逻辑,并根据数据分析结果做出最佳决策和预测。目前机器学习已大量应用于各行各业,助力各产业或科学研究的深度提升。美国加州斯坦福大学的Tony Wyss-Coray团队于2022年6月在Nature Reviews Genetics上发表的一篇名为“Measuring biolo...阅读... 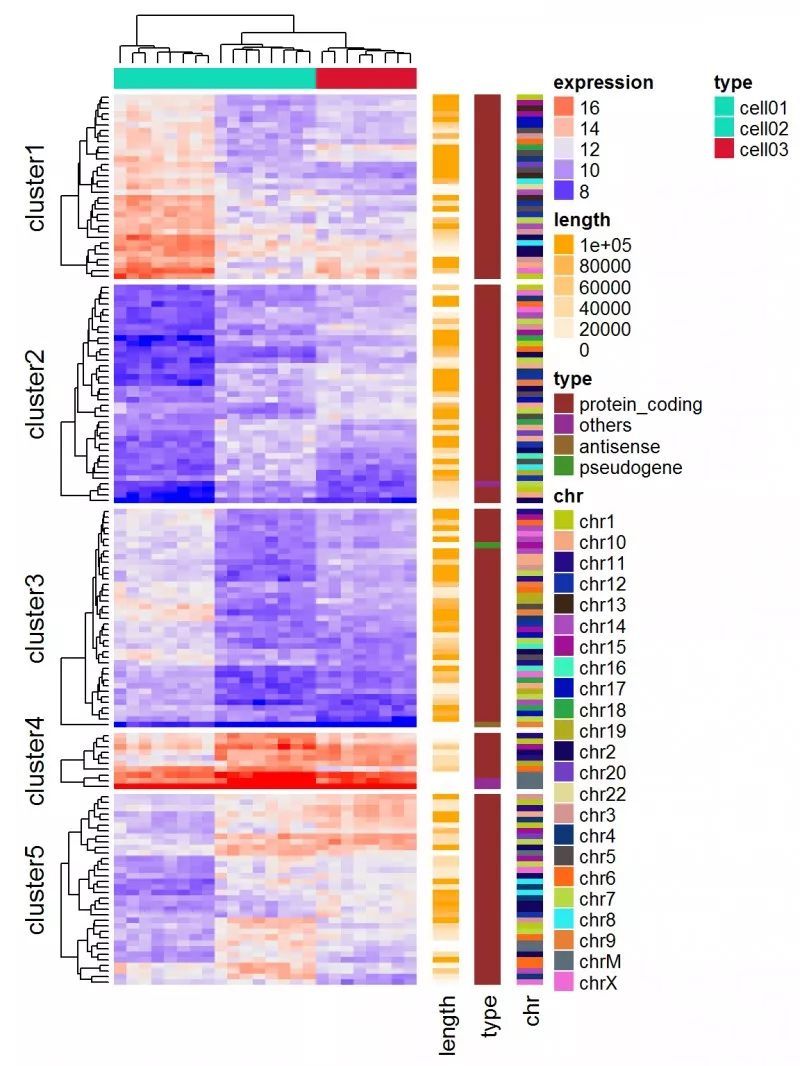 作者简介taoyan:R语言中文社区特约作家,伪码农,R语言爱好者,爱开源。个人博客: https://ytlogos.github.io/简介本文将绘制静态与交互式热图,需要使用到以下R包和函数:heatmap():用于绘制简单热图的函数heatmap.2():绘制增强热图的函数d3heatmap:用于绘制交互式热图的R包ComplexHeatmap:用于绘制...阅读全文>>...
作者简介taoyan:R语言中文社区特约作家,伪码农,R语言爱好者,爱开源。个人博客: https://ytlogos.github.io/简介本文将绘制静态与交互式热图,需要使用到以下R包和函数:heatmap():用于绘制简单热图的函数heatmap.2():绘制增强热图的函数d3heatmap:用于绘制交互式热图的R包ComplexHeatmap:用于绘制...阅读全文>>...  往期我们学习过一个表达趋势折线图+模块热图+GO富集注释的组合类图表绘制,详情可戳:《炫一个模块热图+GO功能富集组合图展示空间功能特征!》 ,后期图表美化主要使用AI完成,如下图。这两天看到另一个比较有意思的R包ClusterGVis,可以同时完成聚类+富集,并生成发表级的组合图表,效果如下。非常方便省事,适合懒人。下面就来学习一番!#相关R包下载与载入:devtools::install_gi...
往期我们学习过一个表达趋势折线图+模块热图+GO富集注释的组合类图表绘制,详情可戳:《炫一个模块热图+GO功能富集组合图展示空间功能特征!》 ,后期图表美化主要使用AI完成,如下图。这两天看到另一个比较有意思的R包ClusterGVis,可以同时完成聚类+富集,并生成发表级的组合图表,效果如下。非常方便省事,适合懒人。下面就来学习一番!#相关R包下载与载入:devtools::install_gi...  正常情况下,我们绘制的常规热图都是直直的,这样更便于我们进行数据的展示和阅读;但上周收到小伙伴的问题:能不能把热图“卷”起来,绘制环状的热图?当然也是可以的,我们先看几个文献中的案例:( BMC Genomics ,2018) ( Microbial Biotechnology , 2017) ( Cell ,2021) 环状热图我们也经常会在论文中看到,用法和热图相同,但更适合于需要展示较多基因...
正常情况下,我们绘制的常规热图都是直直的,这样更便于我们进行数据的展示和阅读;但上周收到小伙伴的问题:能不能把热图“卷”起来,绘制环状的热图?当然也是可以的,我们先看几个文献中的案例:( BMC Genomics ,2018) ( Microbial Biotechnology , 2017) ( Cell ,2021) 环状热图我们也经常会在论文中看到,用法和热图相同,但更适合于需要展示较多基因... 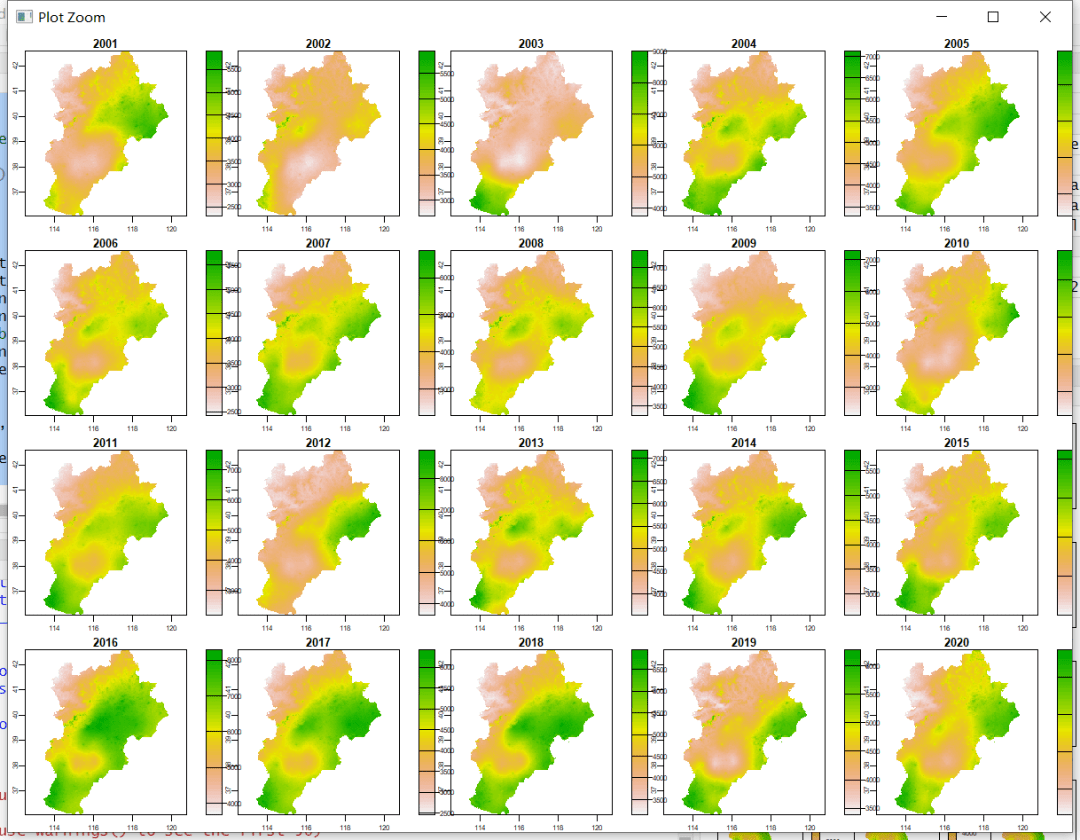 R语言栅格时间序列回归分析——整体和逐像元计算,并行计算前面给大家分享了 2001-2020年1km分辨率中国降水量数据 ,有同学问如何做回归分析,接下来以一元线性回归举例,从整体上和逐像元两种方法来计算。使用的R语言程序包terra 包,栅格计算,支持并行计算tidyverse 包,R语言数据处理可视化必备,内含 ggplot2ggpmis...阅读全文>>...
R语言栅格时间序列回归分析——整体和逐像元计算,并行计算前面给大家分享了 2001-2020年1km分辨率中国降水量数据 ,有同学问如何做回归分析,接下来以一元线性回归举例,从整体上和逐像元两种方法来计算。使用的R语言程序包terra 包,栅格计算,支持并行计算tidyverse 包,R语言数据处理可视化必备,内含 ggplot2ggpmis...阅读全文>>...  大家好,这里是专注表观组学十余年,领跑多组学科研服务的易基因。在全球范围内,牛为60多亿人提供了重要的营养来源。传染病是养牛生产的主要限制因素,且许多疾病人畜共患,因此与人类健康直接相关。近年来牛的许多复杂性状遗传基础得到了广泛研究,但不同动物的大量重要表型变化仍无法解释,人们越来越关注非遗传变异(包括基因表达、DNA甲基化和染色质)与重要牛表型的潜在相关性。为充分了解牛性状的非遗...阅读全文&...
大家好,这里是专注表观组学十余年,领跑多组学科研服务的易基因。在全球范围内,牛为60多亿人提供了重要的营养来源。传染病是养牛生产的主要限制因素,且许多疾病人畜共患,因此与人类健康直接相关。近年来牛的许多复杂性状遗传基础得到了广泛研究,但不同动物的大量重要表型变化仍无法解释,人们越来越关注非遗传变异(包括基因表达、DNA甲基化和染色质)与重要牛表型的潜在相关性。为充分了解牛性状的非遗...阅读全文&...  生信分析类研究,可以说是近两年火热的科研话题了。随着大数据时代的来临,「拍脑门」想选题的时代已一去不复返,从「别人的数据中」挖掘出自己所需的研究核心已经是大势所趋。更何况,据传说——做生信分析的人可以「不做实验」、「无需编程」、「坐在电脑前」、「轻松发文章」。哇!想想都能笑出声来……忍不住暗搓搓的摩拳擦掌一番,打开一篇生信文章,一窥其真容,看看能不能从...阅读全文>>...
生信分析类研究,可以说是近两年火热的科研话题了。随着大数据时代的来临,「拍脑门」想选题的时代已一去不复返,从「别人的数据中」挖掘出自己所需的研究核心已经是大势所趋。更何况,据传说——做生信分析的人可以「不做实验」、「无需编程」、「坐在电脑前」、「轻松发文章」。哇!想想都能笑出声来……忍不住暗搓搓的摩拳擦掌一番,打开一篇生信文章,一窥其真容,看看能不能从...阅读全文>>... 