2024
11-20
11-20
基因序列到进化树:轻松掌握多物种DNA序列比对与进化分析
 基因序列到进化树:轻松掌握多物种DNA序列比对与进化分析基因序列的多物种比对与进化分析是研究物种间遗传关系和功能保守性等的关键步骤。今天,我们将以T2R41基因为例,手把手教大家如何下载基因序列、进行多序列比对,以及构建分子进化树。1. 搜索基因并下载序列首先,打开NCBI官网[1],在搜索栏输入目标基因名称T2R41。输入名称进入搜索结果页面后,...阅读全文>>... 阅 读 全 部 >
基因序列到进化树:轻松掌握多物种DNA序列比对与进化分析基因序列的多物种比对与进化分析是研究物种间遗传关系和功能保守性等的关键步骤。今天,我们将以T2R41基因为例,手把手教大家如何下载基因序列、进行多序列比对,以及构建分子进化树。1. 搜索基因并下载序列首先,打开NCBI官网[1],在搜索栏输入目标基因名称T2R41。输入名称进入搜索结果页面后,...阅读全文>>... 阅 读 全 部 >
 多序列比对(Multiple Sequence Alignment, MSA),对多个序列进行对位排列。这通常需要保证序列间的等同位点处在同一列上,并通过引进小横线(-)以保证最终的序列具有相同的长度。在生物信息分析中,我们有时需要进行多序列比对,Biopython可以帮我们实现,特别是使用linux系统的同学,biopython值得拥有。两种读取方法Biopython提供了两种方法读取多序......
多序列比对(Multiple Sequence Alignment, MSA),对多个序列进行对位排列。这通常需要保证序列间的等同位点处在同一列上,并通过引进小横线(-)以保证最终的序列具有相同的长度。在生物信息分析中,我们有时需要进行多序列比对,Biopython可以帮我们实现,特别是使用linux系统的同学,biopython值得拥有。两种读取方法Biopython提供了两种方法读取多序...... 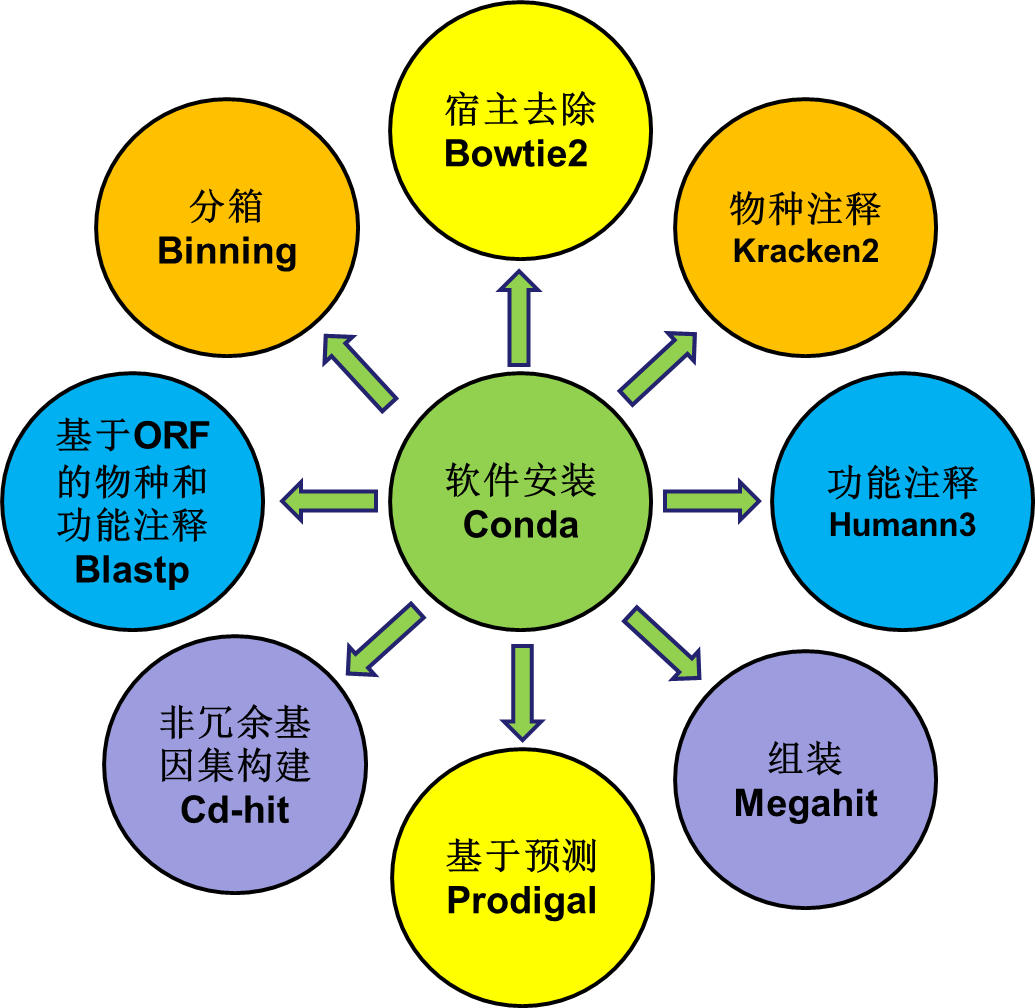 随着高通量技术的发展,宏基因组学(metagenomics)已经成为研究微生物群落物种及功能的前沿科学,在肠道微生物、环境微生物等研究领域具有广泛应用。宏基因组学通过对微生物群落全部DNA进行高通量测序,将测序序列与公共数据库进行比对或从头组装出微生物基因组,从而识别微生物群落的物种和功能基因。目前主流的宏基因组数据分析方法包括三种:基于测序结果进行组装的分析方法;基于reads直...阅读全文&...
随着高通量技术的发展,宏基因组学(metagenomics)已经成为研究微生物群落物种及功能的前沿科学,在肠道微生物、环境微生物等研究领域具有广泛应用。宏基因组学通过对微生物群落全部DNA进行高通量测序,将测序序列与公共数据库进行比对或从头组装出微生物基因组,从而识别微生物群落的物种和功能基因。目前主流的宏基因组数据分析方法包括三种:基于测序结果进行组装的分析方法;基于reads直...阅读全文&... 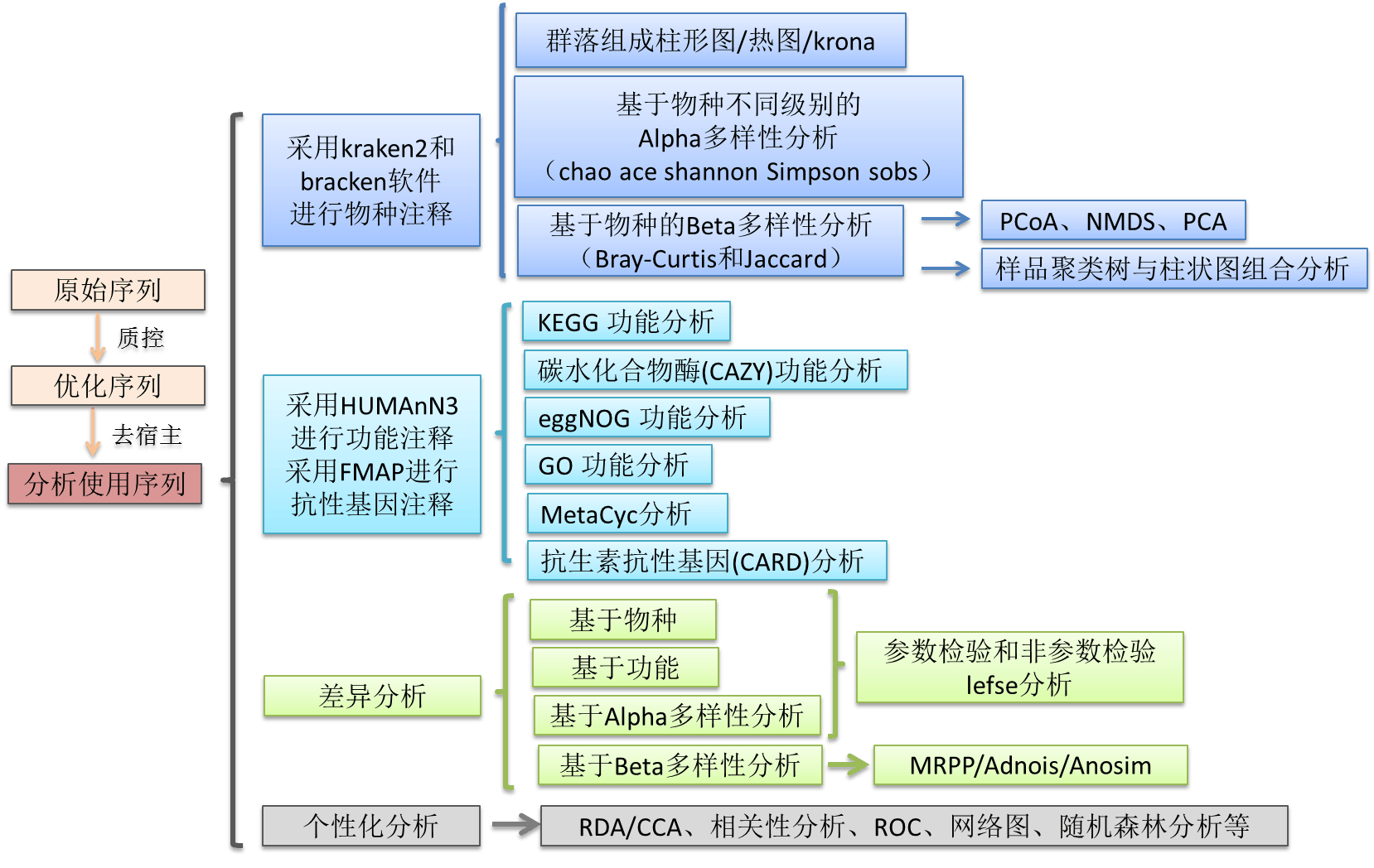 一、 介绍宏基因组 ( Metagenome) 指特定环境下所有生物遗传物质的总和。它包含了可培养的和未可培养的微生物的基因。一般从环境样品中提取基因组DNA, 进行高通量测序,从而分析微生物多样性、种群结构、功能信息、与环境之间的关系等。宏基因组的分析目前主要包括三种方法:基于组装分析、基于reads分析、基于bin分析。下面我们介绍基于Reads比对的分析方...阅读全文>>...
一、 介绍宏基因组 ( Metagenome) 指特定环境下所有生物遗传物质的总和。它包含了可培养的和未可培养的微生物的基因。一般从环境样品中提取基因组DNA, 进行高通量测序,从而分析微生物多样性、种群结构、功能信息、与环境之间的关系等。宏基因组的分析目前主要包括三种方法:基于组装分析、基于reads分析、基于bin分析。下面我们介绍基于Reads比对的分析方...阅读全文>>...  高通量序列比对文件一般可以使用服务器查看,但是如果没有服务器,我们该如何查看呢?这时,就该Tablet软件出场了!操作步骤我们首先下载安装好Tablet软件首先,我们准备好高通量序列比对的结果文件,可以是以下格式。BAMSAMACEAFGMAQSOAPAligner这里,我们以BAM文件为例,首先,点击open这里,我们准备好BAM文件和基因组注释GTF文件打开后,可以看到CONTIG界面,我们可...
高通量序列比对文件一般可以使用服务器查看,但是如果没有服务器,我们该如何查看呢?这时,就该Tablet软件出场了!操作步骤我们首先下载安装好Tablet软件首先,我们准备好高通量序列比对的结果文件,可以是以下格式。BAMSAMACEAFGMAQSOAPAligner这里,我们以BAM文件为例,首先,点击open这里,我们准备好BAM文件和基因组注释GTF文件打开后,可以看到CONTIG界面,我们可...  上一期为大家介绍了用主流基因组数据库查询物种基因组的办法。那么问题来了,当在这些数据库中没有查到目标物种参考基因组,就一定是无参吗?这期我们就给大家列举一些主流基因组数据库之外的Genome Data base。JGI基因组数据库以植物基因组为主,且植物基因组更新较快,其他物种的更新较慢。1、植物:https://phytozome.jgi.doe.gov/pz/po...阅读全文>>...
上一期为大家介绍了用主流基因组数据库查询物种基因组的办法。那么问题来了,当在这些数据库中没有查到目标物种参考基因组,就一定是无参吗?这期我们就给大家列举一些主流基因组数据库之外的Genome Data base。JGI基因组数据库以植物基因组为主,且植物基因组更新较快,其他物种的更新较慢。1、植物:https://phytozome.jgi.doe.gov/pz/po...阅读全文>>...  所研究物种是否有可用参考基因组是做转录组测序之前需要确认的第一个问题,这决定了我们是做有参转录组测序还是无参转录组测序。有参转录组项目会将测到的转录本序列直接比对到参考基因组,以确定基因信息;无参的话,我们就需要借助拼接软件,获得unigene集后去做注释来获得基因信息。基因组只有序列信息可以做为转录组测序的参考基因组吗?答案是不能哦!转录组测序的参考基因组不仅需要有序列信息,还要...阅读全文&...
所研究物种是否有可用参考基因组是做转录组测序之前需要确认的第一个问题,这决定了我们是做有参转录组测序还是无参转录组测序。有参转录组项目会将测到的转录本序列直接比对到参考基因组,以确定基因信息;无参的话,我们就需要借助拼接软件,获得unigene集后去做注释来获得基因信息。基因组只有序列信息可以做为转录组测序的参考基因组吗?答案是不能哦!转录组测序的参考基因组不仅需要有序列信息,还要...阅读全文&... 