2022
06-28
06-28
如何绘制新颖好看的火山图?
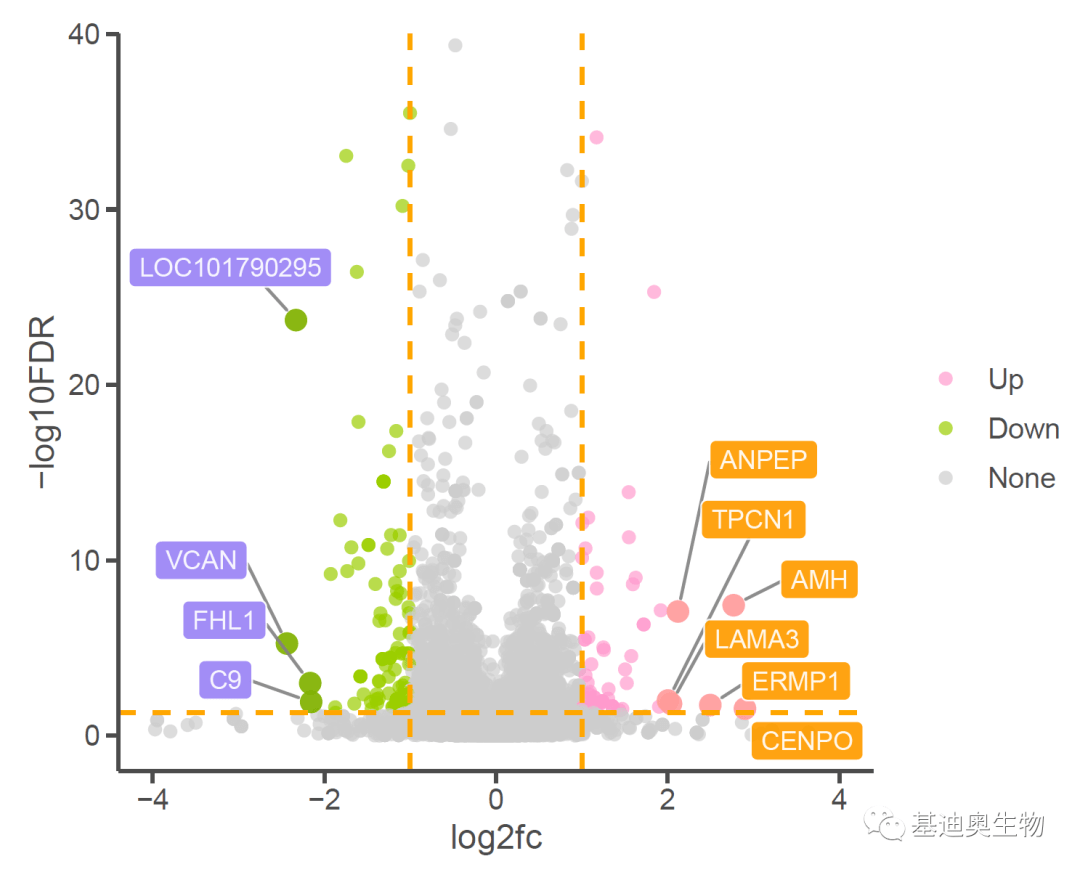 火山图(Volcano plot)是一种比较“远古”的一种散点图,广泛应用于转录组、蛋白组等组间差异分析结果的展示。图表的横轴一般展示差异倍数的变化,而纵轴表示差异分析结果的可靠性。我这里对传统的火山图做了进一步的优化,比如通过调整点的大小突出展示感兴趣基因对应的点,并将感兴趣基因的名称以标签的方式展示出来,如下。那么,如何绘制这般个性又好看的火山图呢?我这里主要用到的还是最常见的ggplot2包... 阅 读 全 部 >
火山图(Volcano plot)是一种比较“远古”的一种散点图,广泛应用于转录组、蛋白组等组间差异分析结果的展示。图表的横轴一般展示差异倍数的变化,而纵轴表示差异分析结果的可靠性。我这里对传统的火山图做了进一步的优化,比如通过调整点的大小突出展示感兴趣基因对应的点,并将感兴趣基因的名称以标签的方式展示出来,如下。那么,如何绘制这般个性又好看的火山图呢?我这里主要用到的还是最常见的ggplot2包... 阅 读 全 部 >
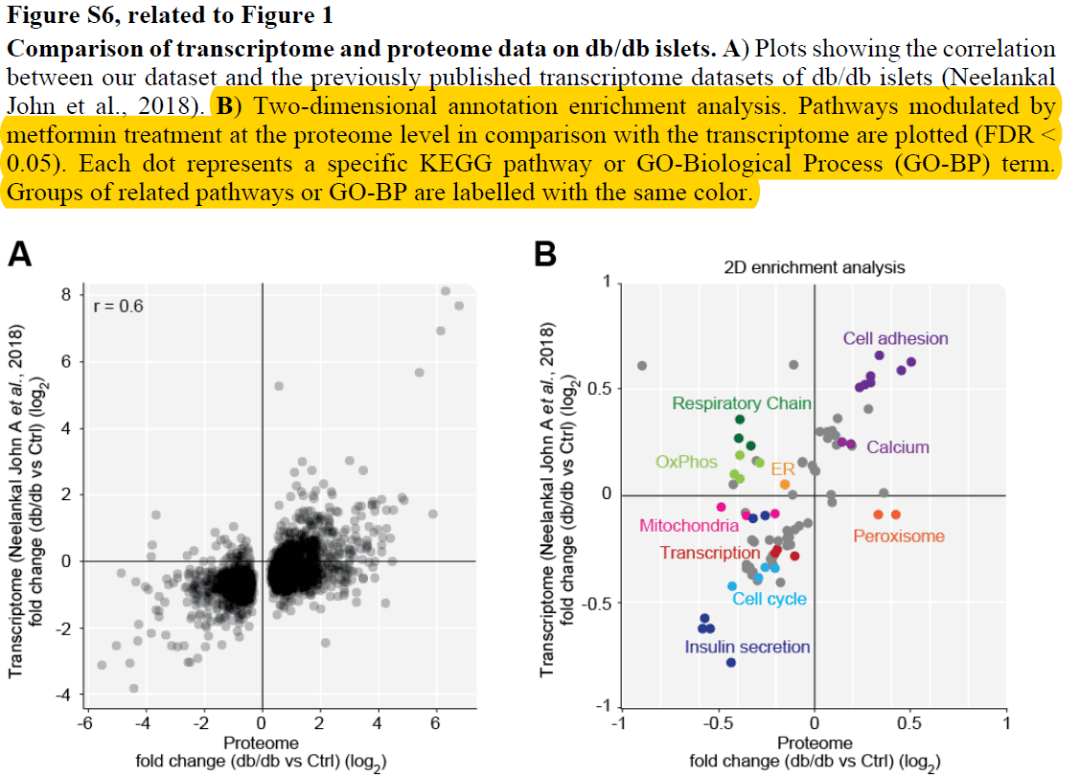 使用Perseus软件进行1D和2D annotation enrichment分析某天,小编在某篇蛋白组学文献中看到这么一个分析,称为2D annotation enrichment的方法。作者使用2D annotation enrichment比较了自己的蛋白组数据集和先前研究中蛋白组数据集的蛋白表达的差异,并将这种差异关联到富集的通路上进行比较,以阐述二者的一致性和区别。图B是2D...阅读...
使用Perseus软件进行1D和2D annotation enrichment分析某天,小编在某篇蛋白组学文献中看到这么一个分析,称为2D annotation enrichment的方法。作者使用2D annotation enrichment比较了自己的蛋白组数据集和先前研究中蛋白组数据集的蛋白表达的差异,并将这种差异关联到富集的通路上进行比较,以阐述二者的一致性和区别。图B是2D...阅读...  作者:严涛 浙江大学作物遗传育种在读研究生(生物信息学方向) 伪码农,R语言爱好者,爱开源编者按: 数据可视化 是解析、理解和展示数据不可缺少的一部分。炫或不炫看个人喜好和功底,能否达意是最基本的要求---最合适的图示和配色表达最直观的含义。长文多图预警,这是关于ggplot2使用的极详细教程(190+图),是入门和晋级参考的不二手册。 前面部分是关于qplot的使用,后面是 ggplot2......
作者:严涛 浙江大学作物遗传育种在读研究生(生物信息学方向) 伪码农,R语言爱好者,爱开源编者按: 数据可视化 是解析、理解和展示数据不可缺少的一部分。炫或不炫看个人喜好和功底,能否达意是最基本的要求---最合适的图示和配色表达最直观的含义。长文多图预警,这是关于ggplot2使用的极详细教程(190+图),是入门和晋级参考的不二手册。 前面部分是关于qplot的使用,后面是 ggplot2...... 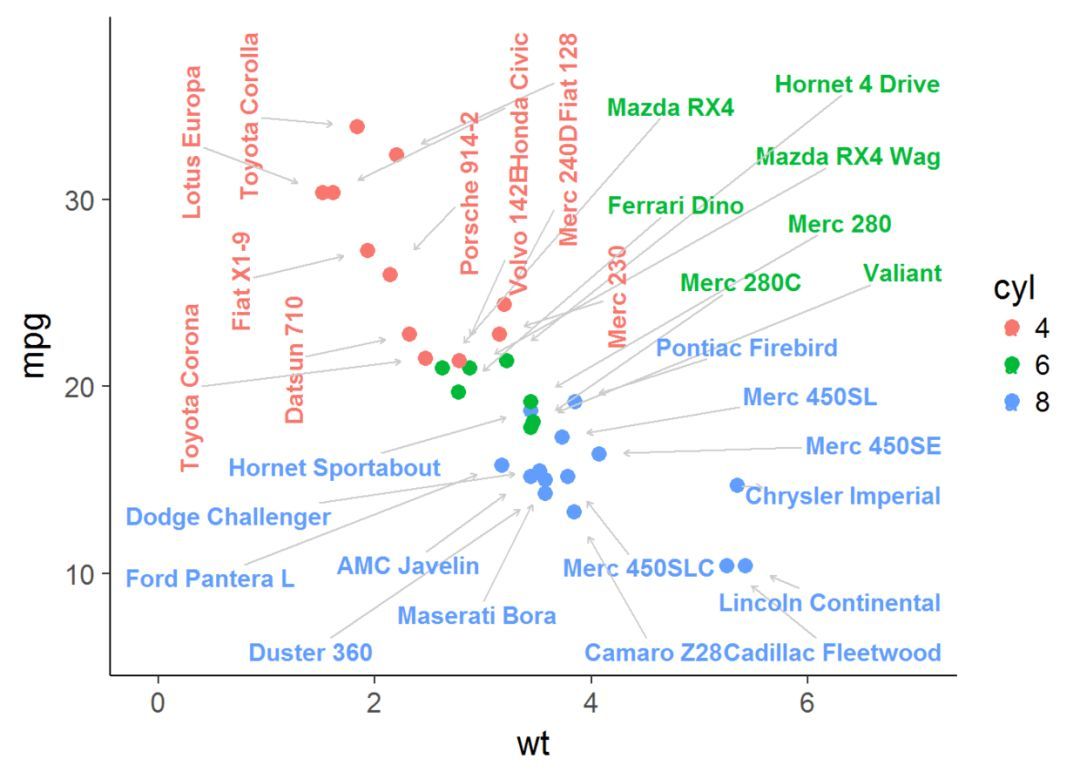 taoyan:伪码农,R语言爱好者,爱开源。个人博客: https://ytlogos.github.io/library(ggplot2)#使用数据集mtcars演示ggplot(mtcars)+ geom_point(aes(wt, mpg), color="red")+geom_text(aes(wt, mpg, label=rownames(mtcars)))+theme_cl...阅读全...
taoyan:伪码农,R语言爱好者,爱开源。个人博客: https://ytlogos.github.io/library(ggplot2)#使用数据集mtcars演示ggplot(mtcars)+ geom_point(aes(wt, mpg), color="red")+geom_text(aes(wt, mpg, label=rownames(mtcars)))+theme_cl...阅读全...  昨天我们介绍了一些网络分析当中用到的一些基础的知识(相互作用网络分析基础 )。对于基因组数据分析而言的话,我们能用到网络分析的就是蛋白相互作用分析(protein-protein ineraction, PPI)分析了。蛋白相互作用分析的数据库有很多,至于为什么选择STRING,还是在于其强大的可视化,以及自定义功能。这样我们可以得到数据结果的同时,还可以得到相对好看的图。下面我们就来介绍一下ST...
昨天我们介绍了一些网络分析当中用到的一些基础的知识(相互作用网络分析基础 )。对于基因组数据分析而言的话,我们能用到网络分析的就是蛋白相互作用分析(protein-protein ineraction, PPI)分析了。蛋白相互作用分析的数据库有很多,至于为什么选择STRING,还是在于其强大的可视化,以及自定义功能。这样我们可以得到数据结果的同时,还可以得到相对好看的图。下面我们就来介绍一下ST...  你还在为找不到课题方案而焦虑吗,复现一篇论文吧!你还在为找不到研究思路而苦恼吗,复现一篇论文吧!你还在为找不到数据方法而悔恨吗,复现一篇论文吧预计阅读时间4分钟。。。。。刚来的小伙伴不要慌了哈,小助理选的一直是同一篇论文只不过用的是不同方法进行复现一千种方法练一次不如一种方法练一千次进步的第一步就是敲下...阅读全文>>...
你还在为找不到课题方案而焦虑吗,复现一篇论文吧!你还在为找不到研究思路而苦恼吗,复现一篇论文吧!你还在为找不到数据方法而悔恨吗,复现一篇论文吧预计阅读时间4分钟。。。。。刚来的小伙伴不要慌了哈,小助理选的一直是同一篇论文只不过用的是不同方法进行复现一千种方法练一次不如一种方法练一千次进步的第一步就是敲下...阅读全文>>... 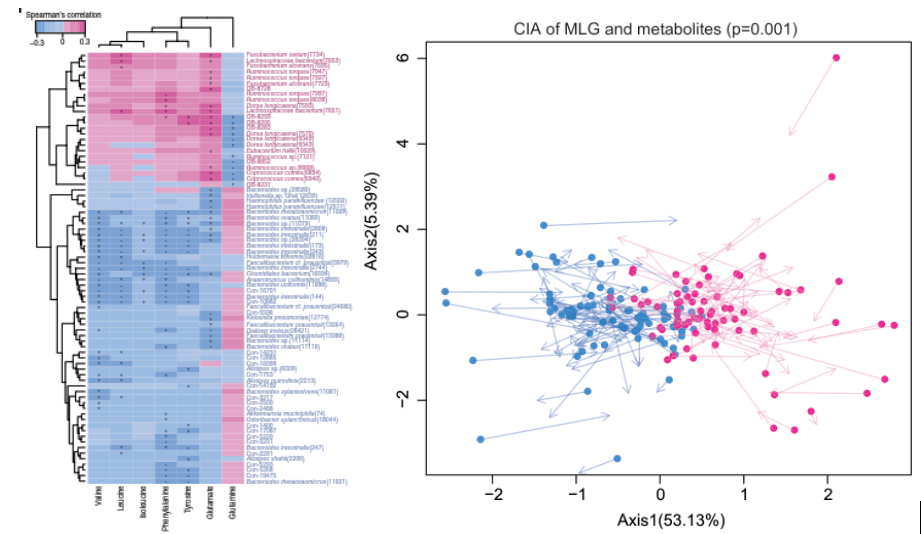 微生物组测序 (主要指扩增子测序、全长扩增子测序与宏基因组测序)可提供细菌构成、基因丰度和功能性信息,可以解决“who is there”(那儿有谁)和“what are they doing”(在干嘛)的问题。而代谢组学是研究生物体中代谢产物变化的科学,可以解决“what have really happened”(究竟发生了什么)的问题。生物科学研究过程复杂,单独和片面的单一组学无...阅读全...
微生物组测序 (主要指扩增子测序、全长扩增子测序与宏基因组测序)可提供细菌构成、基因丰度和功能性信息,可以解决“who is there”(那儿有谁)和“what are they doing”(在干嘛)的问题。而代谢组学是研究生物体中代谢产物变化的科学,可以解决“what have really happened”(究竟发生了什么)的问题。生物科学研究过程复杂,单独和片面的单一组学无...阅读全... 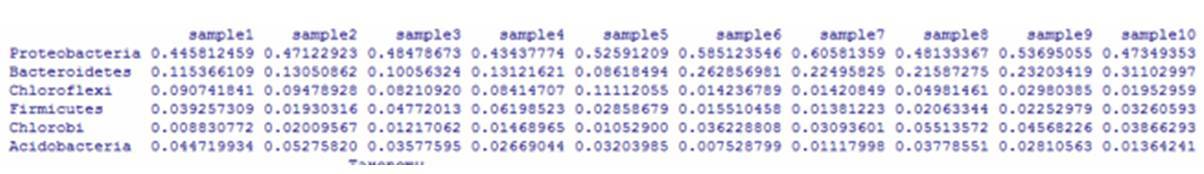 前几期我们介绍了扩增子测序分析流程,包括:微生物群落组成分析,α多样性分析,β多样性分析。接下来几期将结合R语言将扩增子测序分析结果可视化,从而更直观地展现群落结构及丰度分布。R是一门用于统计计算和作图的语言,其强大的绘图功能主要由众多的图形函数来实现,包括:高级绘图函数,低级绘图函数,图形参数。本期主要介绍通过柱状图,热图,韦恩图实现对微生物群落组成分析的可视化。1. 柱状图输入文件:在OTU聚...
前几期我们介绍了扩增子测序分析流程,包括:微生物群落组成分析,α多样性分析,β多样性分析。接下来几期将结合R语言将扩增子测序分析结果可视化,从而更直观地展现群落结构及丰度分布。R是一门用于统计计算和作图的语言,其强大的绘图功能主要由众多的图形函数来实现,包括:高级绘图函数,低级绘图函数,图形参数。本期主要介绍通过柱状图,热图,韦恩图实现对微生物群落组成分析的可视化。1. 柱状图输入文件:在OTU聚...  来源:解螺旋·医生科研助手导语我们把筛出来的差异表用一种直观的图表示出来,一般使用热图(heatmap)将差异表达基因进行数据可视化处理,传统的方法采用R语言包里面的(heatmap)函数对其进行绘制,这里重点讲解一下heatmap包各个常用参数的使用,如果要求较高可以采用这种方法来画图。另外,如果要求不高,也可以使用Heatmap builder这个软件做图,窗口化操作,方法简单。...阅读全文...
来源:解螺旋·医生科研助手导语我们把筛出来的差异表用一种直观的图表示出来,一般使用热图(heatmap)将差异表达基因进行数据可视化处理,传统的方法采用R语言包里面的(heatmap)函数对其进行绘制,这里重点讲解一下heatmap包各个常用参数的使用,如果要求较高可以采用这种方法来画图。另外,如果要求不高,也可以使用Heatmap builder这个软件做图,窗口化操作,方法简单。...阅读全文...  热图是科研论文中一种常见的可视化手段,因其丰富的色彩变化和饱满的信息涵盖量,往往是一篇文章中最引人注目的所在之一。上至 CNS 顶刊,下至1到 2分小文,可以说热图无处不在,堪称文章C位。如何做出一张完美的热图,是居家旅行(科研写作),拜访亲朋好友(征服 editor和reviewer 的心)必备技能。本次教程,我们将为大家详细讲述如何使用R语言绘制高大上的热图。什么是热图...阅读全文>&...
热图是科研论文中一种常见的可视化手段,因其丰富的色彩变化和饱满的信息涵盖量,往往是一篇文章中最引人注目的所在之一。上至 CNS 顶刊,下至1到 2分小文,可以说热图无处不在,堪称文章C位。如何做出一张完美的热图,是居家旅行(科研写作),拜访亲朋好友(征服 editor和reviewer 的心)必备技能。本次教程,我们将为大家详细讲述如何使用R语言绘制高大上的热图。什么是热图...阅读全文>&... 