2023
04-08
04-08
全基因组关联分析(GWAS) — 群体结构
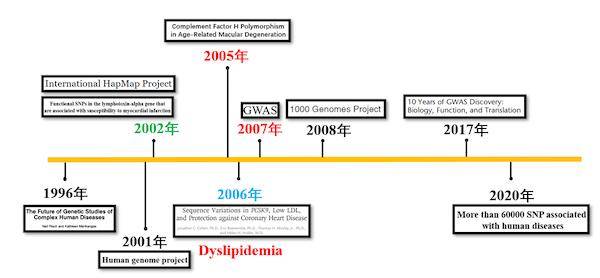 全基因组关联分析(GWAS)目前已经成为研究复杂性状和疾病遗传变异的有效手段,但是由于群体结构的存在,导致分析结果出现假阳性。经过数十年的发展,新的方法的不断出现,才使得群体结构对分析的影响进一步降低。Timeline of GWAS目前GWAS主要采取两种实验设计,一种是基于情缘关系的群体(Population-based cohorts)设计;另一种是基于无关个体的病例-对照(...阅读全文&... 阅 读 全 部 >
全基因组关联分析(GWAS)目前已经成为研究复杂性状和疾病遗传变异的有效手段,但是由于群体结构的存在,导致分析结果出现假阳性。经过数十年的发展,新的方法的不断出现,才使得群体结构对分析的影响进一步降低。Timeline of GWAS目前GWAS主要采取两种实验设计,一种是基于情缘关系的群体(Population-based cohorts)设计;另一种是基于无关个体的病例-对照(...阅读全文&... 阅 读 全 部 >
 细胞器基因组研究相对于微生物基因组来说,要相对容易的多,因为细胞器基因组不大(高等植物叶绿体基因组大小基本在130~160kb左右,动物线粒体基因组基本都在20kb以内),拷贝数高,GC含量与基因组有较大差别(针对大部分物种),所以在高通量测序技术应用非常普通的今天,细胞器基因组很容易从total DNA测序数据中拼接分离出来。而且,细胞器基因组的基因构成相对很稳定,所以细胞器基因组研究的初步工作...
细胞器基因组研究相对于微生物基因组来说,要相对容易的多,因为细胞器基因组不大(高等植物叶绿体基因组大小基本在130~160kb左右,动物线粒体基因组基本都在20kb以内),拷贝数高,GC含量与基因组有较大差别(针对大部分物种),所以在高通量测序技术应用非常普通的今天,细胞器基因组很容易从total DNA测序数据中拼接分离出来。而且,细胞器基因组的基因构成相对很稳定,所以细胞器基因组研究的初步工作...  DNA在染色体上是高度折叠的,DNA与DNA片段之间不可避免的形成了高强度的交互作用。最先提出的3C(Chromosome Conformation Capture)技术,用于测定染色体特定位点之间的交互作用。之后发展出了4C、5C 技术, 分别用于测定染色体上一点到多点和多点与多点之间的交互作用。在2009年Job Dekker 又开发出了Hi-C 技术实现了全基因组范围内的染色体片段间的相互作...
DNA在染色体上是高度折叠的,DNA与DNA片段之间不可避免的形成了高强度的交互作用。最先提出的3C(Chromosome Conformation Capture)技术,用于测定染色体特定位点之间的交互作用。之后发展出了4C、5C 技术, 分别用于测定染色体上一点到多点和多点与多点之间的交互作用。在2009年Job Dekker 又开发出了Hi-C 技术实现了全基因组范围内的染色体片段间的相互作...  2018年5月8日,中国农业科学院棉花研究所所长李付广研究员、武汉大学朱玉贤院士及中棉所杜雄明研究员与北京百迈客生物科技有限公司合作的亚洲棉基因组文章发表在NatureGenetics上,该研究是以全新的亚洲棉基因组为基础,进行遗传进化和全基因组关联分析,对亚洲棉的品质、产量和抗病性等重要性状进行了研究。以下是对该文章的详细解读。1.摘要亚洲棉(Gossypium arboreum)和草棉(......
2018年5月8日,中国农业科学院棉花研究所所长李付广研究员、武汉大学朱玉贤院士及中棉所杜雄明研究员与北京百迈客生物科技有限公司合作的亚洲棉基因组文章发表在NatureGenetics上,该研究是以全新的亚洲棉基因组为基础,进行遗传进化和全基因组关联分析,对亚洲棉的品质、产量和抗病性等重要性状进行了研究。以下是对该文章的详细解读。1.摘要亚洲棉(Gossypium arboreum)和草棉(...... 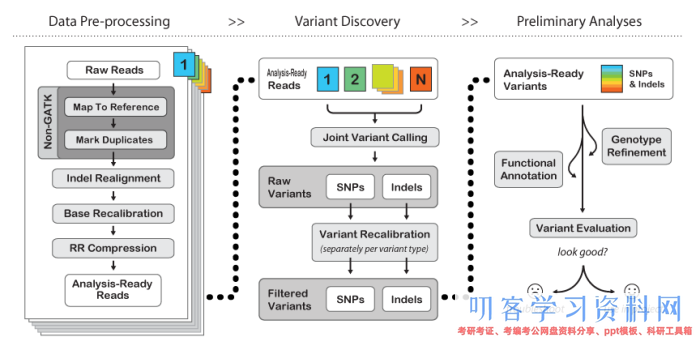 首先说说GATK可以做什么。它主要用于从sequencing 数据中进行variant calling,包括SNP、INDEL。比如现在风行的exome sequencing找variant,一般通过BWA+GATK的pipeline进行数据分析。要run GATK,首先得了解它的网站(http://www.broadinstitute.org/gatk/)。一.准备工作...阅读全文>&g...
首先说说GATK可以做什么。它主要用于从sequencing 数据中进行variant calling,包括SNP、INDEL。比如现在风行的exome sequencing找variant,一般通过BWA+GATK的pipeline进行数据分析。要run GATK,首先得了解它的网站(http://www.broadinstitute.org/gatk/)。一.准备工作...阅读全文>&g...  研究表明,几乎所有生物的表型变异,环境适应和物种形成都与基因组间的结构变异有关。人类基因组既包括蛋白质编码基因,也包括控制这些基因何时表达以及表达到何种程度的调控信息。结构变异(StructureVariantions,简称SVs)是造成物种表型差异的一个重要原因,且与各类疾病,特别是癌症的发生、发展紧密相关,因此研究结构变异非常重要。基因组结构变异通常是指长度大于1Kb的基因组序列变异,包......
研究表明,几乎所有生物的表型变异,环境适应和物种形成都与基因组间的结构变异有关。人类基因组既包括蛋白质编码基因,也包括控制这些基因何时表达以及表达到何种程度的调控信息。结构变异(StructureVariantions,简称SVs)是造成物种表型差异的一个重要原因,且与各类疾病,特别是癌症的发生、发展紧密相关,因此研究结构变异非常重要。基因组结构变异通常是指长度大于1Kb的基因组序列变异,包...... 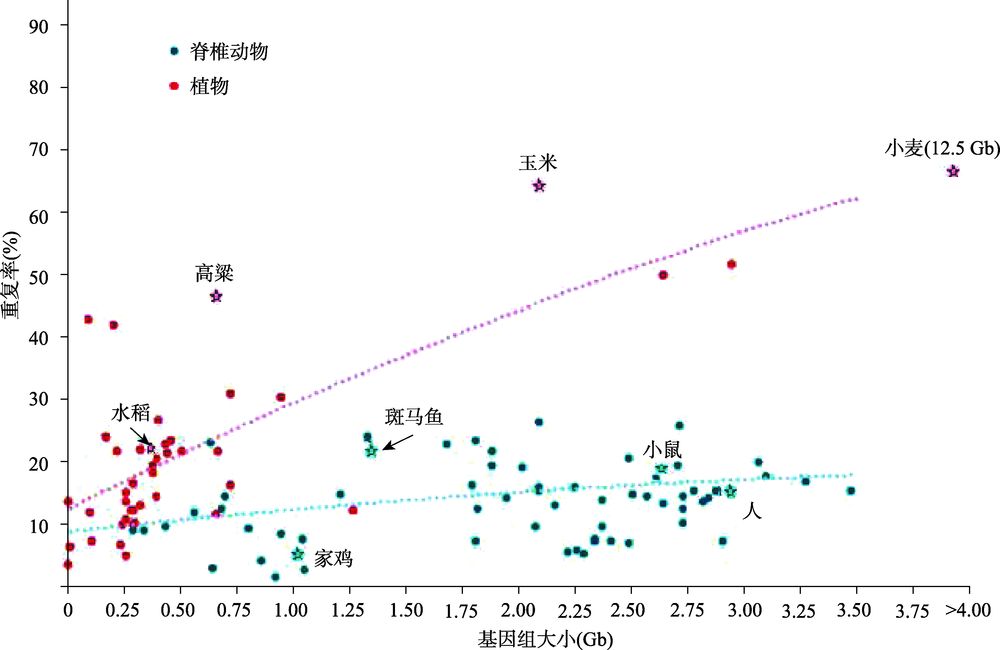 复杂基因组测序技术研究进展内容来自遗传, 2018, 40(11): 944-963 doi: 10.16288/j.yczz.18-255高胜寒, 禹海英, 吴双阳, 王森, 耿佳宁, 骆迎峰, 胡松年中国科学院北京基因组研究所中国科学院基因组科学与信息重点实验室, 北京 100101摘要:复杂基因组指的是无法使用常规测序和组装手段直接解析的一...阅读全文>>...
复杂基因组测序技术研究进展内容来自遗传, 2018, 40(11): 944-963 doi: 10.16288/j.yczz.18-255高胜寒, 禹海英, 吴双阳, 王森, 耿佳宁, 骆迎峰, 胡松年中国科学院北京基因组研究所中国科学院基因组科学与信息重点实验室, 北京 100101摘要:复杂基因组指的是无法使用常规测序和组装手段直接解析的一...阅读全文>>...  开源 Python 和命令行程序 gget 可以高效、轻松地以编程方式访问存储在各种大型公共基因组参考数据库中的信息。 gget 与可获取用户生成的测序数据的现有工具一起使用 ,以取代在基因组数据分析过程中效率低下、可能容易出错的手动网络查询。虽然 gget 模块的灵感来自于繁琐的单细胞 RNA-seq 数据分析任务),但我们预计它们可用于广泛的生物信息学任务。gget文章...阅读全文>&...
开源 Python 和命令行程序 gget 可以高效、轻松地以编程方式访问存储在各种大型公共基因组参考数据库中的信息。 gget 与可获取用户生成的测序数据的现有工具一起使用 ,以取代在基因组数据分析过程中效率低下、可能容易出错的手动网络查询。虽然 gget 模块的灵感来自于繁琐的单细胞 RNA-seq 数据分析任务),但我们预计它们可用于广泛的生物信息学任务。gget文章...阅读全文>&...  川芎Ligusticum chuanxiong Hort.是伞形科藁本属草本植物,以干燥的根茎入药,始载于《神农本草经》, 已有1500多年的种植历史,是我国传统的大宗中药材。川芎广泛分布于我国四川彭州、什邡、眉山等地,在云南、贵州、广西、湖北、江西等省也少量引进种植。川芎性温,味辛、微苦,具有活血行气、祛风止痛的功效,其主要化学成分包含挥发油、生物碱、有机酸和多糖等,在临床上广泛地用于治疗心脏病...
川芎Ligusticum chuanxiong Hort.是伞形科藁本属草本植物,以干燥的根茎入药,始载于《神农本草经》, 已有1500多年的种植历史,是我国传统的大宗中药材。川芎广泛分布于我国四川彭州、什邡、眉山等地,在云南、贵州、广西、湖北、江西等省也少量引进种植。川芎性温,味辛、微苦,具有活血行气、祛风止痛的功效,其主要化学成分包含挥发油、生物碱、有机酸和多糖等,在临床上广泛地用于治疗心脏病...  随着测序成本的不断降低,测序通量的不断提升,基因组大数据时代已经到来。无论是拟南芥的 1001 基因组研究项目,还是 3000 株水稻基因组的公布,都让研究者们获得了大量可用的测序数据,同时,不同品系的动植物基因组也逐渐发布,如拟南芥的 col 和 Ler,水稻的 9311 和 Nipponbare,那么如何快速的比较两套不同品系的参考基因组,从而准确鉴别两者之间的差异 SNP &a...阅读全文...
随着测序成本的不断降低,测序通量的不断提升,基因组大数据时代已经到来。无论是拟南芥的 1001 基因组研究项目,还是 3000 株水稻基因组的公布,都让研究者们获得了大量可用的测序数据,同时,不同品系的动植物基因组也逐渐发布,如拟南芥的 col 和 Ler,水稻的 9311 和 Nipponbare,那么如何快速的比较两套不同品系的参考基因组,从而准确鉴别两者之间的差异 SNP &a...阅读全文... 