
2018年5月8日,中国农业科学院棉花研究所所长李付广研究员、武汉大学朱玉贤院士及中棉所杜雄明研究员与北京百迈客生物科技有限公司合作的亚洲棉基因组文章发表在Nature Genetics上,该研究是以全新的亚洲棉基因组为基础,进行遗传进化和全基因组关联分析,对亚洲棉的品质、产量和抗病性等重要性状进行了研究。以下是对该文章的详细解读。
 1.摘要
1.摘要
亚洲棉(Gossypium arboreum)和草棉(Gossypium herbaceum)的祖先是现代栽培异源四倍体棉花A亚基因组的供体。本研究中通过整合了不同的技术,提升了亚洲棉的基因组组装水平;同时对243株二倍体棉花(亚洲棉和草棉)进行全基因组重测序分析,绘制基因组变异图谱,并发现亚洲棉和草棉(A)与雷蒙德氏棉(D)同时进行了分化;单独的对亚洲棉分析表明亚洲棉起源于中国南部,随后被引入长江和黄河地区,大多数具有驯化相关特性的种质都经历了地理隔离;通过亚洲棉的全基因组关联分析(GWAS),鉴定了亚洲棉11个重要农艺性状的98个显著关联位点,GaKASIII的非同义替换(半胱氨酸/精氨酸替换)使得棉籽中的脂肪酸组成(C16:0和C16:1)发生了变化;棉花枯萎病抗性与GaGSTF9基因的表达激活相关。本研究对理解棉花A亚基因组的进化具有重要的意义。
2. 研究背景
棉花是世界上最重要的商业作物之一,同时也是研究植物多倍化的有价值的资源。亚洲棉最可能在马达加斯加或印度河流域文明(巴基斯坦摩亨佐达罗)开始驯化,随后分散到非洲和亚洲一些地区。亚洲棉最初在1000多年前作为观赏植物引入中国。当在地方的农业生态环境适应和人类选择影响的过程中,中国的Gossypium arboreum形成了独特的地理种群,称之为“sinense cotton”。
虽然棉花种植者已经基于RFLP和SSR markers构建了各种遗传图谱,但是G. arboreum和G. herbaceum优良农艺和经济性状的基因尚未被鉴定。同样通过种内和种间特异性杂交将二倍体的重要特性引入四倍体并不是富有成效的,G. raimondii,G. arboreum,G. hirsutum和G. barbadense基因组序列的发布为研究群体遗传,栽培和驯化提供了先决条件。通过GWAS和QTLs在水稻,玉米,大豆,谷子,黄瓜,番茄和陆地棉中鉴定了许多候选基因。本研究中,利用了三代PacBio和Hi-C技术,重新组装了高质量的亚洲棉基因组,分析了243份二倍体棉花种质的群体结构和基因组分化趋势,同时确定了一些有助于棉花皮棉产量遗传改良的候选基因位点。
3. 材料和方法
1、测序材料:
基因组测序材料:二倍体G. arboreum栽培品种cultivar Shixiya1(SXY1);
自然群体材料选择:243份棉花,包含230份亚洲棉 G. arboretum 和13份草棉 G. herbaceum [243份棉花选自国家种质基因库(中国安阳),种植在中国农业科学院棉花研究所(ICR,CAAS)的温室中],插入片段长度500 bp;测序深度6X;
遗传群体材料选择:亲本(GA0146和GA0149),测序深度20X;2个混池(F2群体,有绒型和无绒型各20个子代),测序深度30X;
群体材料表型调查:在230份亚洲棉中选择了215份表型稳定的材料,分别在河南安阳,海南三亚和新疆阿克苏进行种植,大部分性状选自多年多点的表型数据进行调查,每个地点设置3次重复。
2、测序平台:
PacBio RSII和Illumina HiSeq 2500
3、相关软件:
基因组组装(Canu和Falcon;Quiver;Pbjelly);
TEs转座元件注释(RepeatScout,LTR-FINDER,MITE和PILER;Repbase;REPET;RepeatMasker);
基因预测注释(geMoMa;Augustus;PASA;EVidenceModeler;InterProScan)
群体研究:
比对注释(BWA,Picard,GATK,ANNOVAR);
群体结构分析(FastTree,PHYLIP,STRUCTURE);
连锁不平衡分析(Haploview);
遗传多样性分析(π,Fst);
全基因组关联分析(EMMAX);
4. 研究结果
1. 亚洲棉基因组组装更新
三代+Hi-C:PacBio reads (77.6×);有效Hi-C reads(>20×)
三代组装结果:共计获得了142.54 Gb 原始三代测序数据,组装1.71 Gb亚洲棉基因组,Contig N50=1.1 Mb,最长的Contig为12.37 Mb
(1)Hi-C辅助基因组组装:利用Hi-C技术将组装的1573 Mb的数据定位到13条染色体上,与已经发表的基因组相比,当Hi-C数据比对到更新的基因组后,对角线外的不一致性明显减少(图1 a-b)。
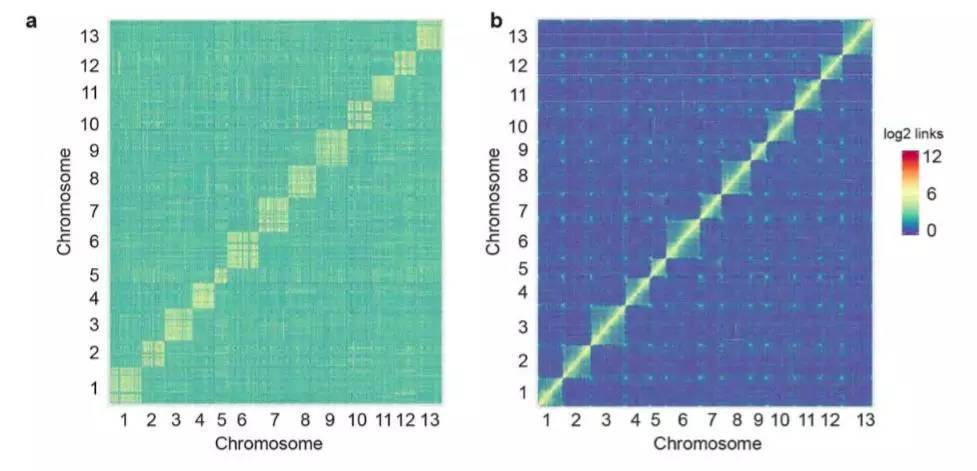
图1 Hi-C数据在两版亚洲棉基因组上的比对
a. Hi-C数据与亚洲棉原基因组比对
b. Hi-C数据与亚洲棉更新基因组比对
(2)基因组共线性分析:进行了亚洲棉A型与异源四倍体陆地棉的AADD型的共线性分析,发现更新后的基因组的共线性更高(图2 a-b)。
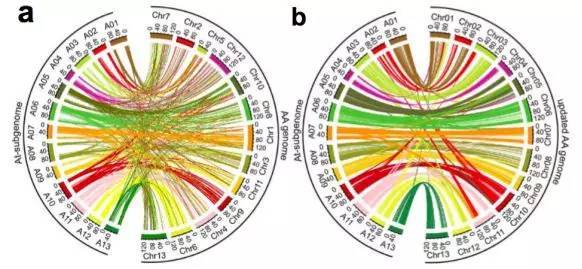
图2 亚洲棉(AA型)与陆地棉(AADD型)共线性分析
a. 亚洲棉原基因组与陆地棉基因组共线性分析;
b. 亚洲棉更新基因组与陆地棉基因组共线性分析
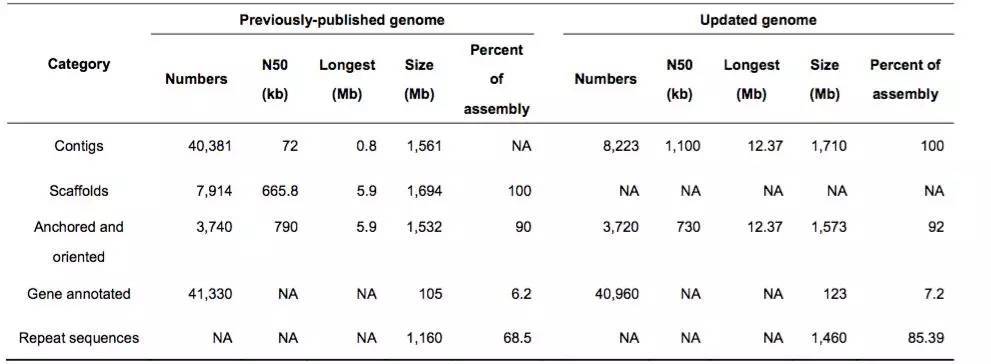
(3)亚洲棉原基因组(二代)与更新后基因组(三代)比较(表1):
表1 亚洲原基因组与更新后基因组的组装指标比较
2. 二倍体棉花群体遗传进化分析
(1)二倍体棉花群体材料选择
共计选择了243份二倍体棉花材料:230份亚洲棉G. arboreum (A2) 和13份草棉G. herbaceum (A1);材料来源:中国南部(SC),长江(YZR)和黄河(这些区域代表了中国二倍体棉花大部分的表型和地理多样性)。
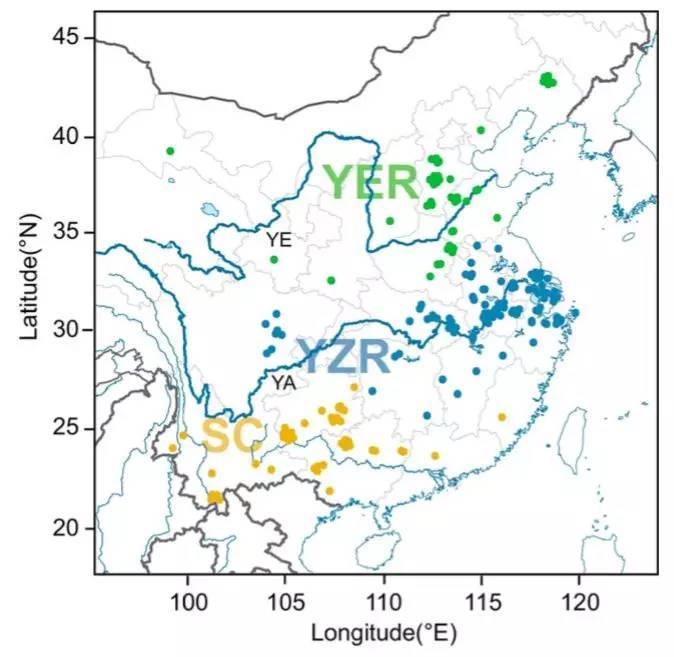
图3 亚洲棉的地理分布
(2)群体重测序数据统计
通过重测序的研究策略,利用Illumina HiSeq 2500 测序平台,PE125双端测序,共计获得了2.29 T数据,平均测序深度~6.0×,以本研究中更新后的亚洲棉基因组为参考基因组进行比对分析,统计获得了17,883,108个高质量SNPs和2,470,515个indels,242,449 个SNPs(1.36%)和16,816个indels(0.68%)位于亚洲棉36,205个基因的编码区。在31,549个基因中,共计鉴定了128,512(0.72%)个非同义突变SNPs,在8,117个基因中共计鉴定了11,372 (0.46%)个indels。
(3)二倍体棉花群体分层分析
以雷蒙德氏棉(G. raimondii)为外群,利用72,419个SNPs构建系统发育树,显示:G. herbaceum(草棉)和G. arboretum(亚洲棉)聚类成2个独立的群(图4 a-b)。G. arboretum(亚洲棉)进一步又分为SC,YZR和YER三个群,显示了地理分布模式的差异,进而利用PCA分析支持这一结果(图4 c)。同时发现了亚洲棉和草棉是由不同的祖先种驯化而形成。
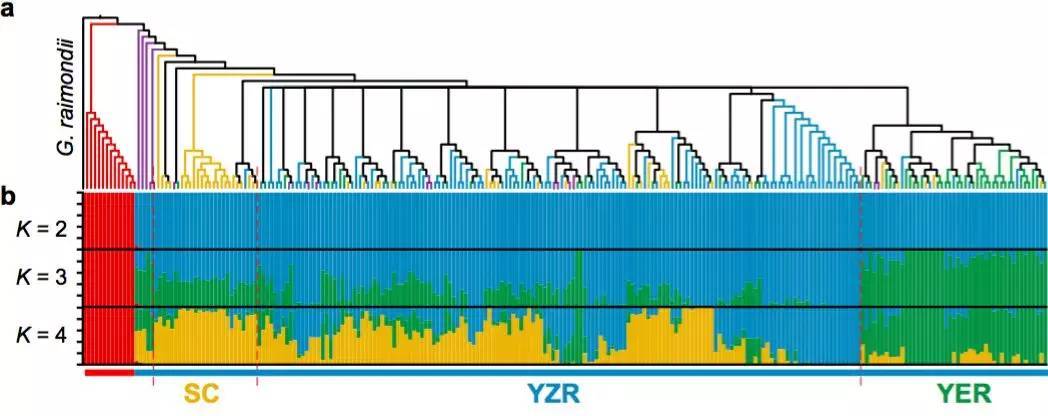
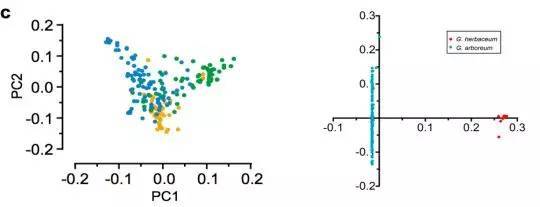
图4 二倍体棉花的群体分层分析
a. 243份二倍体棉花系统发育树
b. 243份二倍体棉花的群体结构分析
c. PCA主成分分析(中国亚洲棉的PCA分析;亚洲棉和草棉的PCA分析)
(4)二倍体棉花LD和选择性清除分析
通过表型计算统计发现,与长江和黄河区域的两个不同地区的材料项目,中国南部的材料的表型相对匮乏。此外中国南部SC的亚洲棉(π=0.211×10-3)比长江流域YZR(π=0.197×10-3)和黄河流域YER(π=0.199×10-3)的亚洲棉的核苷酸多态性高,这表明了亚洲棉最早在中国南部地区种植,并进一步扩展到长江和黄河地区,而这与之前基于SSR分析的结果一致;连锁不平衡分析结果显示,亚洲棉的LD衰减距离约为105.5 kb(r2=0.40),草棉的衰减距离约为145.5 kb(r1=0.39)(图5),其LD衰减距离与大豆(约83 kb)和水稻(籼稻约123 kb;粳稻约167 kb)接近,但远远高于栽培玉米(22-30 kb)。大约有23.9%的亚洲棉和22.9%的草棉的等位基因与雷蒙德氏棉的基因组相一致,暗示了亚洲棉与草棉同时开始分化(图6)。
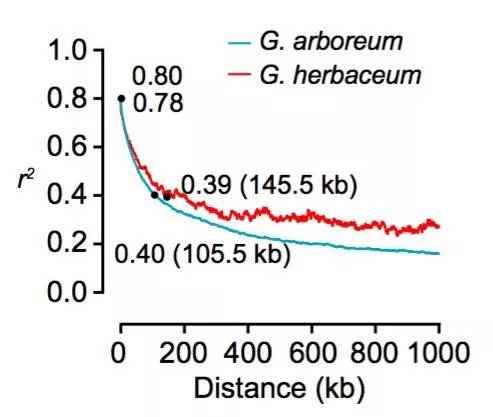
图5 连锁不平衡分析
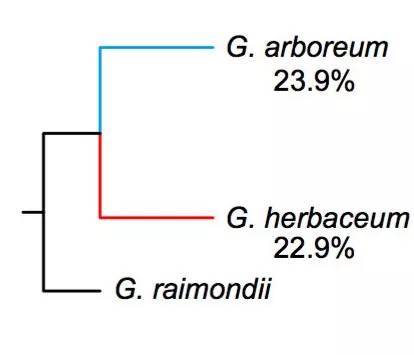
图6 棉属的系统发育与等位基因分析
人工选择在农作物的驯化和迁徙的过程中具有重要的作用。群体结构分析显示当K=4时,YER与SC和YZR明显不同(图4 b,K=4)。通过两两群体间(SC vs. YZR, SC vs. YER, YZR vs. YER)的选择性清除分析(FST)鉴定出了分别覆盖到3,162,2,879和3,308个基因上的59,53和51个显著遗传分化的区域。SC和YZR之间的21个分化的区域(约43.5 Mb 含有915个基因)在群体SC和YER之间是保守的(图7 a)。
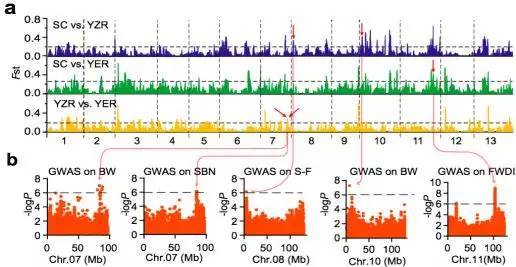
图7 a. 亚洲棉SC,YZR和YER的选择性清除分析
b. 全基因组关联分析
3. 亚洲棉的全基因组关联分析(GWAS)
对来自不同环境下的11个重要性状进行全基因组关联分析,在98个显著关联的信号中,其中25信号个来自基因区(外显子或内含子区),包含与形态性状相关的8个信号区,与产量性状相关的6个信号区,与油籽性状相关的3个信号区;剩余73个信号来自非编码区。大部分农艺性状的GWAS关联信号中显示地理差异,如交配分支数,开花期,铃重和抗病性这些性状定位在保守的基因区(图7 b,表2)。因此推断成熟度,产量和抗病性这些性状一直处于强烈的认为或地理选择之下。
表2 部分与性状关联的SNPs及候选基因
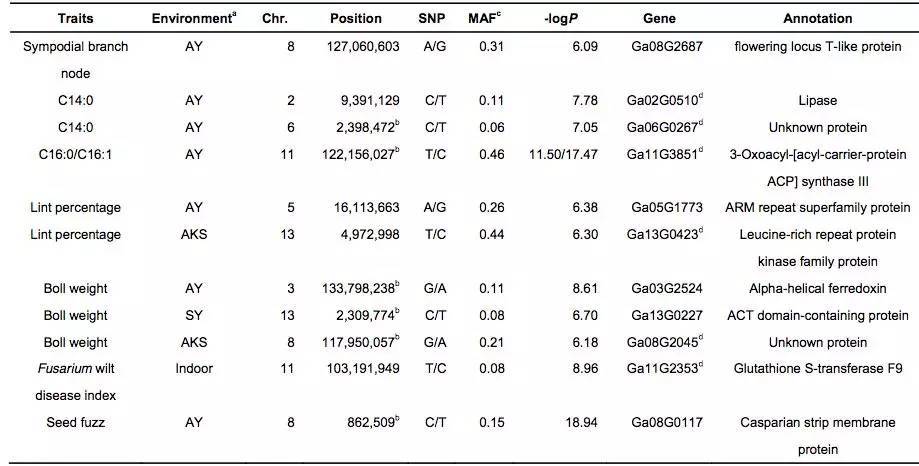
(1)脂肪酸含量相关基因的定位与研究
棉花是世界上第六大植物油来源,通过GWAS关联分析,在11号染色体上的GaKASIII基因组座位上(Ga11G3851)的第8个外显子区获得了1个显著的SNP位点,该基因编码3-Oxoacyl-[acyl-carrier-protein ACP] synthase III(3-氧酰基-[酰载体蛋白]合酶III),如图8 a-c,KASIII基因编码的这一关键酶可以使得脂肪酸链从C2到C4延伸,并最终确定种子中棕榈酸(C16:0)和棕榈油酸(C16:1)的组成。GaKASIII基因的多态性导致了保守的ACP_synthase_III_(酰载体蛋白合酶III)结构域中半胱氨酸/精氨酸间的置换(图8 c),单倍型B(TGT,Cys)主要出现在低含油量种质中,而在高含油量种质中发现单倍型A(CGT,Arg)(图8 d-e)。GaKASIII基因在开花后(DPA)的30天表达量最高(图9),这是种子油量积累的关键阶段,在单倍型种质A中,C16:0和C16:1含量以显着的速率累积(图10);蛋白质结构模型预测显示,半胱氨酸/精氨酸残基位于α螺旋处,该位点靠近酶活性位点,同时是辅酶A(CoA)结合位点(图11)。
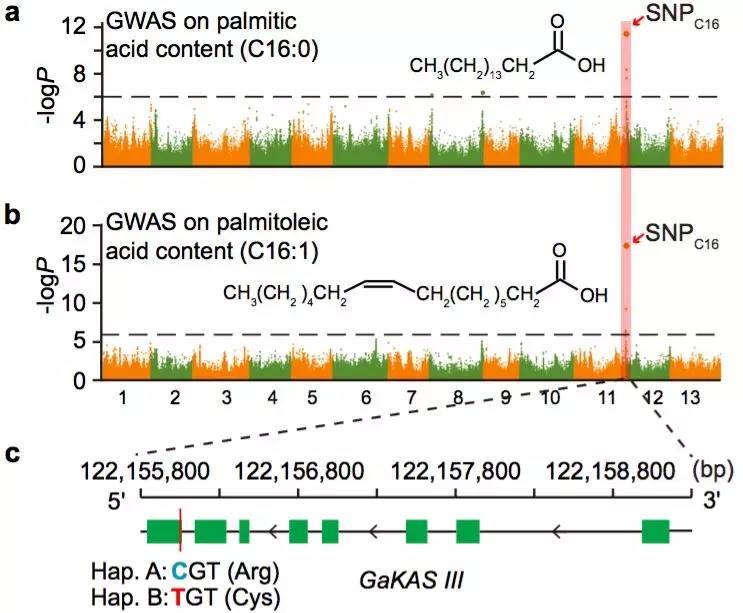
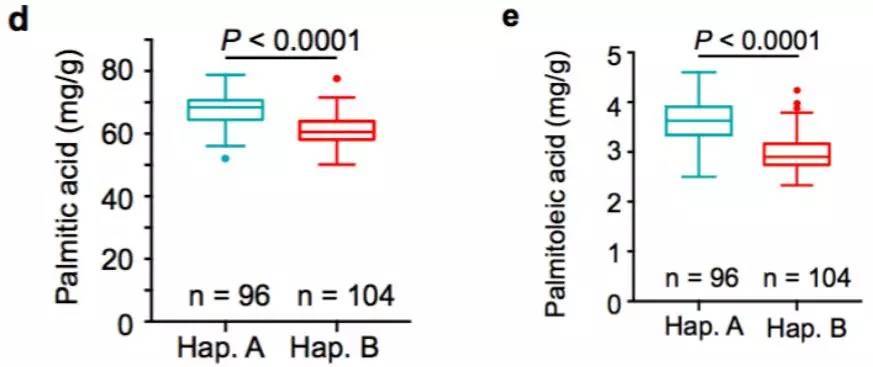
图8 脂肪酸含量GWAS关联分析
a. 棕榈酸含量的GWAS关联分析
b. 棕榈油酸的GWAS关联分析
c. GaKASIII基因的变异(Arg/Cys)
d-e. Hap. A和Hap. B中棕榈酸和棕榈油酸含量比较
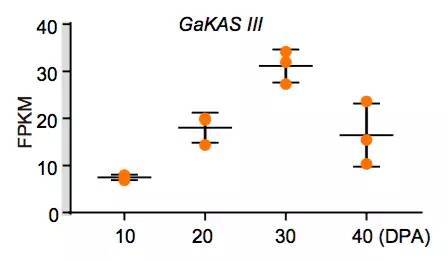
图9 GaKASIII基因在棉花胚珠发育过程中的表达
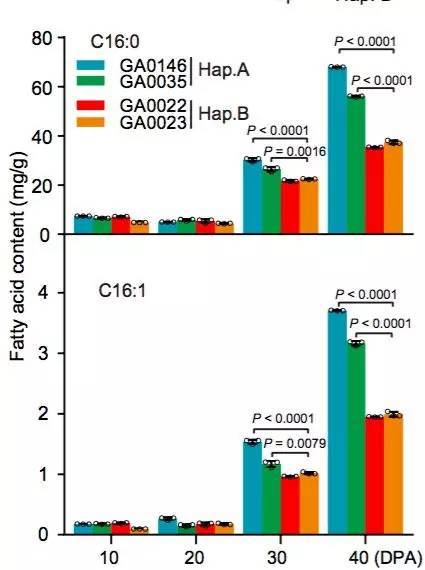
图10 在棉花生长发育过程中C16:0和C16:1脂肪酸含量分析(Hap. A和Hap. B)
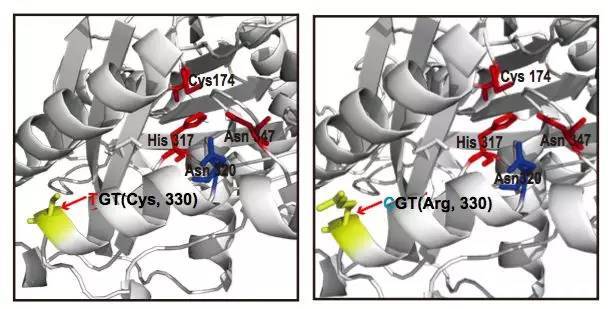
图11 GaKASIII蛋白结构模型
(2)棉花枯萎病抗性相关基因的研究
棉花枯萎病是由尖孢镰刀菌萎蔫专化型Fusarium oxysporum f. sp. vasinfectum (FOV)引起的棉花维管束病害,是棉花产量的重要威胁之一。通过GWAS,进行棉花FOV抗性分析,发现在11号染色体上获得了强的关联信号,其-logP value=8.96(图12 a)。进一步分析表明,关联到的SNP簇位于Ga11G2353基因的上游(图12 b),该基因与拟南芥GSTF9基因为直系同源基因,GSTF9基因编码参与植物对生物和非生物胁迫响应的谷胱甘肽S转移酶(glutathione-S-transferases),携带疾病易感等位基因‘T’(以紫色显示)的种质主要在SC群体中发现,所有YER群体材料携带耐病等位基因‘C’(以橙色显示)(图12 c)。研究发现GSTF9基因仅在FOV接种到亚洲棉幼苗的耐受系中上调(图13),与空载体棉花系(TRV::00)相比,GSTF9基因沉默棉花品系(TRV::GSTF9, the virus-induced gene silencing,VIGS,vector carrying the GSTF9 gene)对于FOV的接种更加敏感(图14-15)。此外,TRV::GSTF9植株系与TRV::00植株系相比,TRV :: GSTF9植株系中的真菌DNA的量显著高于TRV::00植株系,且GST催化活性显着低于TRV::00植株系(图16 a-b),表明GaGSTF9基因可能是亚洲棉FOV抗性的靶标。
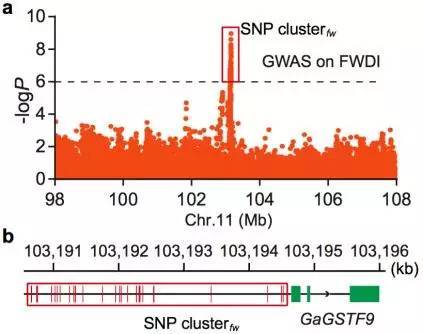
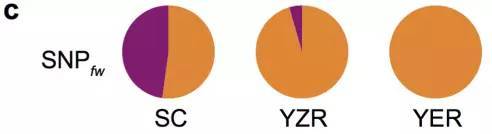
图12 亚洲棉FWDI的全基因组关联分析
a. FWDI的GWAS分析
b. GaGSTF9基因结构及附近关联到的SNPs
c. 关联分析群体基因分型敏感型(紫)、耐受型(橙)
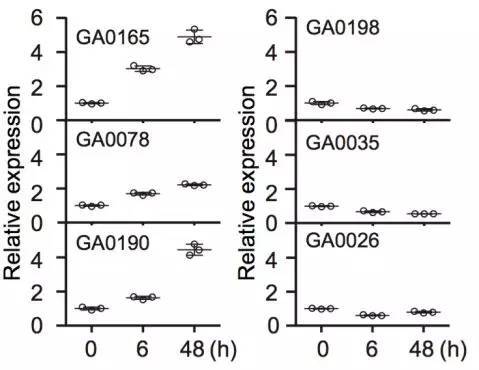
图13 GaGSTF9基因表达的qRT-PCR分析
注:根部接种镰刀霉菌,(高耐受型GA0165,GA0078和GA0190;高敏感型GA0198,GA0035和GA0026)
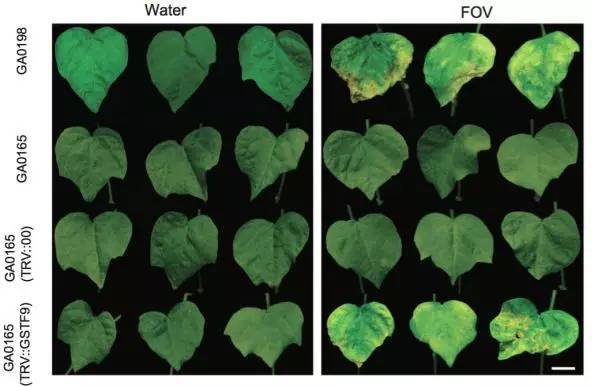
图14 不同植株处理后的病症比较(GA00198,GA0165,TRV::00和TRV::GSTF9,
处理条件:接种水和FOV)
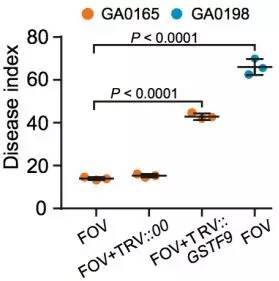
图15 不同植株处理后的疾病感染指数比较(GA00198,GA0165,TRV::00和TRV::GSTF9,处理条件:接种FOV)
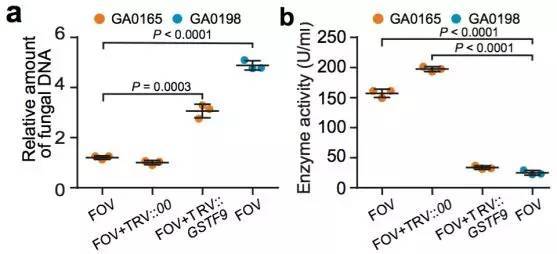
图16 a.GA00198, GA0165, TRV::00和TRV:GSTF9植株中FOV DNA的相对含量测定;
b.GA00198, GA0165, TRV::00和TRV:GSTF9植株中GST酶活性测定;
(3)与棉绒相关基因的研究
棉绒是覆盖种子表面的短纤维。研究中选择了亚洲棉种质中的158份有绒毛和57份无绒毛材料进行GWAS关联分析,最终在8号染色体上(〜0.6 Mb至〜1.3 Mb的区间内)获得了强烈的关联信号(图17 a-b)。QTL分析也同样定位到8号染色体上(图17 c)。通过有绒毛品系(GA0146)和无绒毛品系(GA0149)杂交获得的F2代显示了有绒毛和无绒毛的表型分离比为1:3(图17 d),说明了棉绒的生长是由单基因座控制。研究中进而放大了QTL和GWAS的重叠区,发现了这个大约600 kb的区域包含推测的10个蛋白编码基因(图17 e)。在这一强烈的信号区域(-logP = 18.95)下或附近,发现了4个凯氏带膜蛋白基因(casparian strip membrane protein genes)。在B-型细胞周期蛋白上游获得了1个信号,而B-型细胞周期蛋白之前被报道过与毛状体和纤维发育有关。

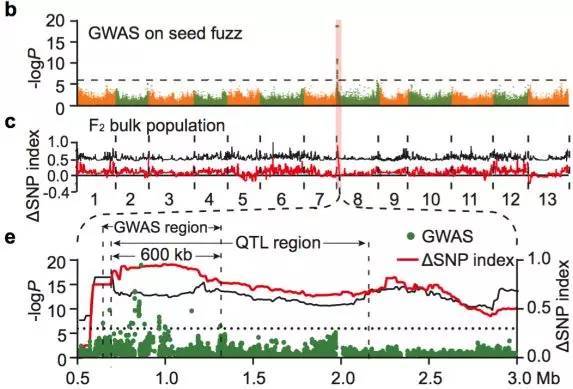
图17 棉绒生长发育基因的联合定位(GWAS+QTL)
a.亚洲棉种子有绒(左)和无绒(右)表型
b.GWAS关联分析;c.F2群体QTL分析
d.F2群体的构建; e.GWAS和QTL联合分析
亚洲棉在中国棉花的栽培史上具有重要的作用。本研究揭示了中国的亚洲棉群体呈现出不同的地理格局,这一观点与其从中国的南部到长江和黄河的引入相一致。几种表型如产量和抗病性状在棉花从中国南部迁徙到长江再到黄河,经历了显著的变化,而这一变化受到了当地环境和人为选择的影响。研究中通过不同种质群体的比较,获得的地理受选择的基因区域与QTL重叠区域是重要的,具高分辨率的遗传资源,这将极大地促进棉花复杂性状的改良。此外,研究中确定了GaKASIII基因可以促进棉花脂肪酸链的延伸和含油量,同时发现了2种典型的GaGSTF9基因单倍型启动子与FOV抗性相关。最后结合GWAS与QTL共同定位的结果,鉴定了凯氏带膜蛋白基因在棉绒细胞的发育过程中可能发挥功能性的作用。本研究表明地理隔离已经影响了SC,YZR和YER群体的遗传基础,同时影响了中国亚洲棉的抗病性和产量性状的形成与分布。
- 本文固定链接: https://maimengkong.com/zixun/1405.html
- 转载请注明: : 萌小白 2023年3月19日 于 卖萌控的博客 发表
- 百度已收录
