
川芎Ligusticum chuanxiong Hort.是伞形科藁本属草本植物,以干燥的根茎入药,始载于《神农本草经》, 已有1500多年的种植历史,是我国传统的大宗中药材。川芎广泛分布于我国四川彭州、什邡、眉山等地,在云南、贵州、广西、湖北、江西等省也少量引进种植。川芎性温,味辛、微苦,具有活血行气、祛风止痛的功效,其主要化学成分包含挥发油、生物碱、有机酸和多糖等,在临床上广泛地用于治疗心脏病、脑梗死及尿路结石等疾病[1-3]。
基因组序列是研究一个物种遗传背景的基础,随着高通量测技术的逐渐成熟,许多植物基因组序列相继被发表,包括大量药用植物基因组。如通过高通量测序技术成功测算出灵芝、丹参、人参、三七、天麻、穿心莲、黄花蒿、广藿香、铁皮石斛等几十种重要药用植物的基因组大小和特征[4-5]。但我国药用植物种类丰富,约占中药材资源总数的87% [6],同大多数药用植物一样,现报道川芎的研究主要集中在化学成分[2]和药理药效机制上[7],分子生物学方面仅开展了川芎转录组分析和利用通用引物分析其遗传多样性[8-10],在分子遗传学系统研究上存在较大空白。虽然药用植物基因测序技术的应用为川芎全基因组测序提供了技术基础,但由于川芎基因组结构庞大,遗传背景复杂,直接进行全基因组测序存在一定困难,因此在进行全基因组测序之前,有必要对川芎基因组大小进行调研。
本研究采用流式细胞术和Illumina Hiseq 2500高通量测序技术相结合的方式对川芎基因组大小进行估算,对所得的基因组数据进行K-mer分析、基因组预测及注释,关注阿魏酸等成分合成途径的基因注释,为进一步全基因组精细测序和药效成分合成分子机制研究提供参考依据和基因资源。
1 材料
分别采集2个月左右的绿豆Vigna radiata (Linn.) Wilczek.、陆地棉Gossypium hirsutum L.幼嫩叶片和1个月左右川芎幼嫩叶片(种植于四川省农科院经济作物育种栽培研究所基地)用于流式细胞分析,并采集川芎叶片用于基因组测序。绿豆由四川省农业科学院经济作物育种栽培研究所叶鹏盛研究员鉴定为豆科豇豆属植物绿豆,陆地棉由中国农业科学院棉花研究所杜雄明研究员鉴定为锦葵科棉属植物陆地棉,川芎由四川农业大学陈兴福教授鉴定为伞形科藁本属植物川芎。
2 方法
2.1 样品的制备
分别选取绿豆、棉花、川芎幼嫩叶片各2份,每份120 mg,洗净置于预冷的培养皿中,向培养皿中加入1 mL预冷的OttoI细胞裂解液,快速切碎叶片后,用移液枪上下吹打混匀(避免气泡),所得的提取液用42 μm尼龙膜滤过到离心管中,低速冷冻离心后,弃上清液,向沉淀中加入1 mL冰浴的OttoII缓冲液重悬细胞,放置4 ℃备用。
采用改良CTAB法提取川芎基因组DNA,使用NanoDrop 2000C超微量分光光度计和1%琼脂糖凝胶电泳检测DNA浓度及完整性。
2.2 流式细胞术测定川芎基因组大小
将上述制备好的细胞悬浮液样品中加50 μL 1 mg/mL RNase,50 μL、50 μg/mL PI(DNA荧光染料碘化丙啶,预先经0.22 μm微孔滤膜滤过,−20 ℃保存),混匀,4 ℃避光染色10 min。随后用FACSCalibur流式细胞仪检测PI在488 nm激发光下发出的荧光,CellQuest软件捕捉荧光信号数据,ModFit软件分析结果。测定基因组大小。
待测样品DNA量=对照DNA量×待测样品的荧光强度/对照品的荧光强度
2.3基因组测序和质量评估
构建270 bp和500 bp的小片段文库,利用Hiseq2500测序技术对文库进行双端测序。从文库中随机取10 000条单端read与NCBI数据库中的核苷酸数据库(NT)进行BLAST [11]比对,判断样本是否被外源物种污染。数据测序由北京百迈客生物科技有限公司完成。
2.4基因组大小、重复序列和杂合率预测
选取K值为21对基因组大小、重复序列、杂合率进行预测。用程序Jellyfish计算K-mer分布,并通过K-mer分布曲线初步评估基因组重复序列含量和杂合度。
基因组大小=K-mer总数/ K -mer 期望深度值
2.5 基因组初步组装和GC-Depth分布分析
利用SOAPdenovo进行组装得到contig,利用双末端信息进行gap填充,将无overlap关系的contig拼接组装成scaffold,获得含有N(重复序列)的初级基因组序列。过滤后的read比对到已组装好的基因序列上,获得碱基深度,以10 kb为窗口,在序列上无重复前进,计算每个窗口的平均深度与GC含量,做出GC_depth图[12]。
2.6 重复序列分析
对测序所得的数据进行重复序列分析。使用4个互补程序LTR_FINDER [13]、MITE-Hunter [14],RepeatScout [15]和PILER-DF [16]构建川芎重复序列文库,随后由PASTEClassifier [17]分类,并与Repbase [18]转座因子库结合起来作为最终的库,然后运行软件Repeat-Masker [19]在最终文库中找到同源重复序列。
2.7 基因预测和注释
基因从头预测,在滤过掉小于1000 bp大小的scaffold后,用Genscan和Augustus软件通过拟南芥的训练集预测川芎基因。将预测到的基因比对到非冗余蛋白序列(non-redundant,Nr)、真核生物蛋白直系同源簇(clusters of euKaryotic orthologous groups,KOG)、基因本体(gene ontology,GO)、swiss-prot和京都基因与基因组百科全书(Kyoto encyclopedia of genes and genomes,KEGG)等数据库进行BLAST分析来对预测基因进行注释。然后,通过KEGG在线网站(http://www.genome. jp/kegg/)检索KEGG途径,使用Blast2GO软件处理得到的Nr注释结果进行GO分类,使用在线网站(http://www.ncbi.nlm.nih. gov/COG/)处理KOG注释结果进行KOG分类。
2.8基因家族鉴定及系统发育树构建
使用ORTHOMCL v2.0.9将预测到的川芎蛋白序列与丹参Salvia miltiorrhizaBunge、胡萝卜Daucus carotavar. sativa Hoffm. 、葡萄Vitis viniferaL.和拟南芥Arabidopsis thalianaL.等4种植物中氨基酸数目大于50的序列汇集到一个蛋白数据库中,通过blastp比对获得所有物种蛋白序列之间的相似性关系,E值为1×10−5,去掉序列一致度小于30%,覆盖率小于30%的序列;并对比对结果进行聚类,默认膨胀系数为1.5。在ORTHOMCL结果中检索单拷贝基因家族,利用单拷贝同源基因基于最大似然法(maximum likelihood,ML)进行进化树构建。
3 结果与分析
3.1流式细胞术检测基因组大小
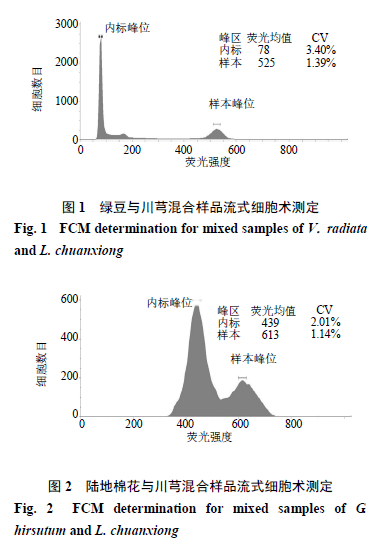
采用绿豆和陆地棉作为内标植物,用流式细胞仪检测其混合样品的荧光强度。已知绿豆基因组大小为579 Mb [20],陆地棉基因组为2173 Mb [21],通过2个内标植物计算出川芎的基因组大小分别为525/78×579=3 897.12 Mb,变异系数(coefficient of variation,CV)为3.40%(图1);613/439×2173=3 034.28 Mb,CV为2.01%(图2)。
3.2 川芎基因组测序数据统计及质量评估
使用川芎基因组DNA构建270 bp的文库,通过Illumina Hiseq2500测序平台测序并过滤得到222.04 Gb高质量的数据,测序深度为72.59 X,测序数据Q20比例均在97.59%以上,Q30比例均在94.74%以上。随机筛选的1000条单端read能够比对上NT核酸数据库的read占总read的7.62%,其中比对上野胡萝Daucus carotaL.、细叶藁本Ligusticum tenuissimum(Nakai) Kitagawa的read数分别占比对上NT库reads数的61.81%、3.01%,且未发现动物、微生物等异常比对,说明样本不存在污染。
3.3 川芎基因组特征
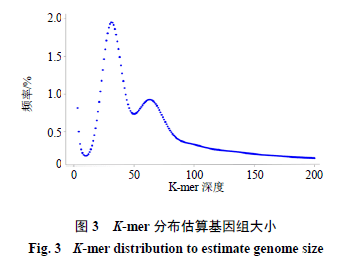
通过270 bp文库数据构建K=21的K-mer分布图(图3),K-mer深度62 X为主峰(由于杂合度较高,本研究对主峰判断参考流式细胞结果),测序得到K-mer的总数为189 618 848 178,估算出川芎基因组大小为3 058.37 Mb,与流式细胞实验中以陆地棉为内标植物测定的结果相近。在主峰对应深度的1/2处出现明显的杂合峰,深度为31 X,说明川芎基因组具有较高的杂合度。进一步根据变异数目占基因组大小的比例即杂合度估算该基因组杂合率,在所有有效数据中检测到每139个碱基对中就有3个SNP,估算出川芎基因组具有较高杂合率,约为2.16%。K-mer分布存在较长拖尾,暗示川芎基因组存在较高的重复率,估算重复序列的基因组大小估计为2 440.13 Mb,约为川芎基因组的79.79%。说明川芎属于高重复、高杂合、大基因组等基因组特征的复杂物种。
3.4川芎基因组组装和GC含量分析
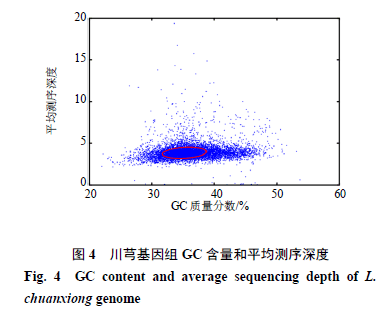
使用222.04 Gb高质量数据,基于K=41组装产生12 973 787个contig和8 119 089个scaffold。contig N50为286 bp,N90为131 bp,最大长度达到26 842 bp,总长度为3 198 598 874 bp;scaffold N50为493 bp,N90为191 bp,最大长度为29 779 bp,总长度为3 284 536 081 bp。其中,contig和scaffold的N50值相对较短,可能是由于川芎高杂合率引起的。通过对组装的contig进行GC含量的统计,结果显示川芎基因组的GC含量约为36.32%(图4),说明测序不具有明显的GC偏向性,不影响测序分析的准确性。
3.5 川芎基因组重复序列分析
重复序列检测显示其总长度为2 250 901 770 bp,约为基因组大小的73.60%,低于K-mer分析估算的重复序列含量,原因可能是组装效果的限制,导致组装过程中重复序列损失6.19%。注释上,能够找到明确重复序列元件的总长度约为2 026.25 Mb。其中,长末端重复序列(long terminal repeated,LTR)是最丰富的重复元件,占基因组的14.14%,其次是长散在重复元件(long interspersed nuclear elements,LINE),占基因组的0.49%。SSR重复序列总长度约49.41 Mb,占基因组的1.62%,占重复序列的2.44%。
3.6 基因预测和注释
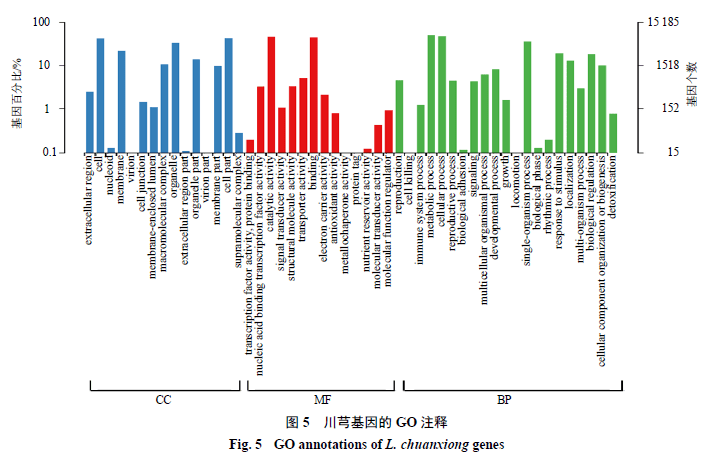
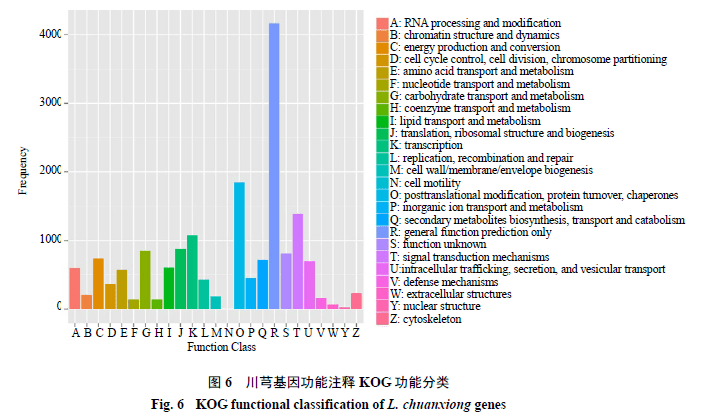
总共预测到34 864个基因,总长度为27 532 625 bp。在预测的基因中,查找到79 130个外显子,总长度为18 557 406 bp;79 129个内含子,总长度为8 975 219 bp。有30 737个基因在功能数据库中能比对到注释信息,Nr具有最高的注释率(30 584,87.72%)KEGG具有最低的注释率(9623,27.60%)。在预测的基因中,15 184个基因被分类为GO功能类别;15 598个基因被分类为KOG功能类别;9623个基因注释到125个KEGG代谢途径。在GO功能类别中,包含分子功能、细胞组成和生物过程(图5);在KOG功能类别中,通用功能预测的基因最多,其次是翻译后修饰、蛋白质周转、分子伴侣和信号转导机制(图6);在参与基因数目最多的前10条KEGG代谢途径中,注释基因分别参与核糖体代谢、植物激素信号转导、内质网蛋白质、剪接体、碳代谢、氨基酸生物合成、RNA转运、氧化磷酸化、植物-病原菌互作、淀粉和蔗糖代谢等途径。
3.7 基因家族鉴定及系统发育分析
通过与胡萝卜、丹参、葡萄和拟南芥等物种蛋白序列比对(图7-A、B),川芎中常见的基因家族中的基因数量小于同科的胡萝卜,每个基因家族的平 均基因数量与其他物种相当,但川芎独特基因家族的数量比其他物种的独特基因家族中的基因数量要大得多,共计 2058个基因家族。所有5个物种共有的基因家族为7112个,其中2001个基因是单拷贝基因,即每个基因家族中只存在一个直系同源基因,可用于系统发育推断和发散时间估计。对2001个单拷贝基因利用ML构建系统发育树(图7-C)。
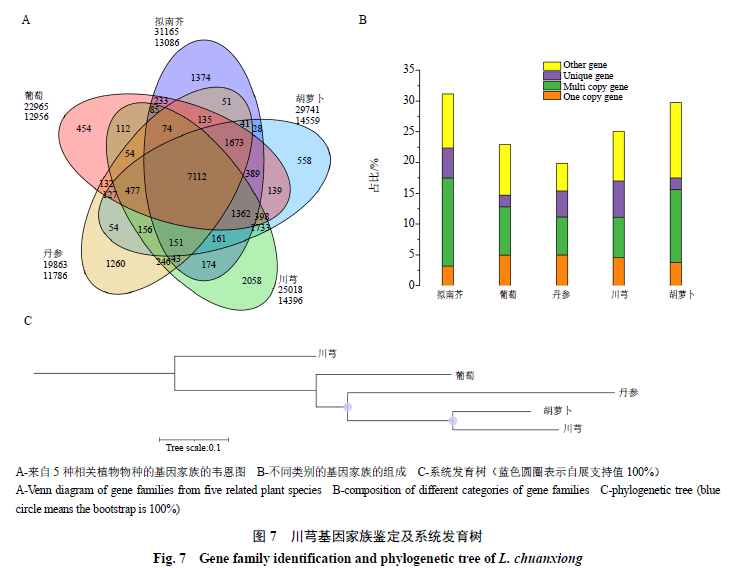
从川芎系统发育树分析,川芎、胡萝卜、丹参、葡萄和拟南芥来自于共同祖先。其中,川芎与胡萝卜最晚与其他物种发生分歧,二者进化分支长度差异最小,分歧时间更短,亲缘关系更近。从遗传变异度上来看,胡萝卜所在的分支最短,遗传变异度最小,进化距离最近,川芎的遗传变异度和进化距离仅次于胡萝卜,丹参的遗传变异度最高,进化距离最远。
3.8参与阿魏酸生物合成的基因
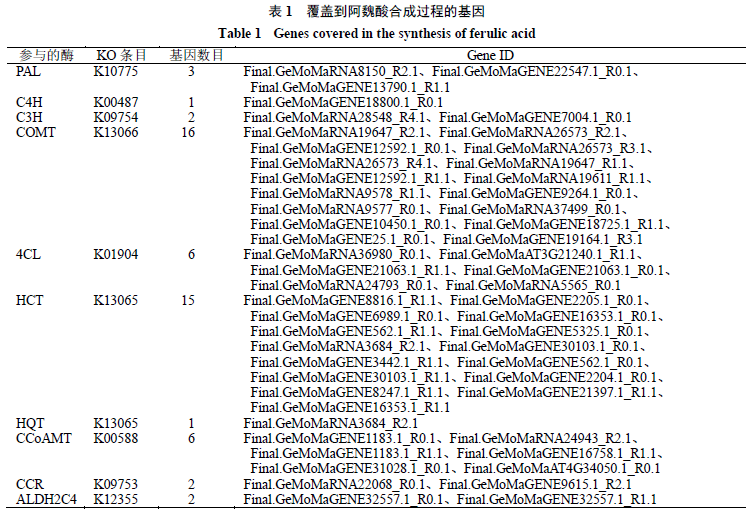
阿魏酸是川芎的主要药效成分,属于苯丙烷生 物合成途径,是木质素合成的中间体。近年来研究者证实了在川芎、当归等伞形科物种中,阿魏酸的合成途径主要包括COMT途径和CCoAMT途径[9,22-24],主要参与的酶有苯丙氨酸解氨酶、肉桂酸-4-羟化酶、香豆酸-3-羟化酶、咖啡酸- O - 甲基转移酶、4-香豆酸辅酶A连接酶、莽草酸O-羟基肉桂酰转移酶、奎宁O-羟基肉桂酰转移酶、咖啡酰辅酶A- O - 甲基转移酶、肉桂酰辅酶A还原酶、醛脱氢酶家族成员C4。为挖掘川芎中参与阿魏酸生物合成的主要基因,本研究从注释的KEGG代谢通路中提取苯丙烷生物合成参考途径(map00940)相关数据,对阿魏酸合成过程的相关基因进行了检索,共有53个功能基因覆盖到阿魏酸合成途径中(表1)。其中,编码咖啡酸- O - 甲基转移酶、4-香豆酸辅酶A连接酶、莽草酸O-羟基肉桂酰转移酶、咖啡酰辅酶A- O - 甲基转移酶等酶的基因均有多个同源拷贝存在,预测川芎基因组在进化的过程中某一时间点发生过基因扩张。
4 讨论
基因组包含了一个生物体所有基因的总和,了解生物体基因组信息有助于深入了解生物体遗传、进化、生物合成、次生代谢等全部过程。通过基因组测序技术可以对特定物种基因组进行测序,利用生物信息学方法对测序序列进行拼接和组装,最终获得该物种基因组序列,进而了解其基因组信息[25]。2009年,陈士林团队[6]首次提出本草基因组计划,此后,越来越多的学者开始在药用植物基因组学研究上投入大量精力。药用植物全基因组水平研究,有助于阐明药用植物活性成分生物合成和代谢调控途径之间的关系,为具有相似有效成分和药理活性的近缘物种间的系统发育关系研究奠定了基础,也为药用植物的遗传育种和基因资源保护提供了重要依据[26]。
本研究利用流式细胞术和高通量测序相结合的方式对川芎基因组进行测算。通过流式细胞术估算 出川芎基因组大小分别为3 897.12 Mb和3 034.28 Mb,通过高通量测序的K-mer分析,综合2个分析结果估算川芎基因组大小为3 058.37 Mb,属于基因组较大的物种。本研究测得川芎基因组GC含量为36.32%,处于植物基因组GC含量应介于25%~65%的合理范围[27],说明川芎基因组测序的结果和组装是正确可靠的。通过K-mer分析,估算出川芎重复序列含量为79.79%,杂合率约为2.16%,与地黄[28]、黄芪[ 29]等药用植物类似,呈现高重复、高杂合的基因组特征,进一步说明川芎基因属于高重复、高杂合、基因组庞大的物种。
在基因预测、注释和基因家族的鉴定中,本研究共预测到川芎编码蛋白基因34 864个,远高于其伞形科亲缘关系较近的胡萝卜(32 113个)[30],可能是因为组装的都是短片段测序文库,川芎中的基因数量可能被高估了。此外,本研究中测序完成后从头组装中产生的contig N50为286 bp,scaffold N50为493 bp,明显较预期短,这与广藿香[4]、罗汉果[12]等药用植物的全基因组调研结果一致。提示对于川芎这种具有复杂基因组特征的物种来说,利用二代高通量测序对其全基因组进行精确测序仍然存在技术难度。因此,提高川芎基因组的测序深度和组装质量,建议后续的研究可采用二代和三代测序技术相结合,并利用全基因组染色体构象捕获技术(high-through chromosome conformation capture,Hi-C),解析全基因组范围内整个染色质DNA在空间位置上的关系,获得完整准确的全基因组图谱[31],得到高质量的川芎基因组序列。
本研究首次利用流式细胞技术和基因组survey分析,初步获得川芎基因组大小和结构特征,即基因组庞大、序列重复率高、序列杂合度高,为下一步进行全基因组精细测序奠定基础。本研究中组装产生的大量川芎基因组序列和注释基因为后续分子标记的开发和基因功能研究提供了大量的资源。同时,本研究挖掘了阿魏酸合成途径中的参与基因,对川芎阿魏酸生物合成途径潜在分子机制的初步研究,为进一步研究川芎生物学和选育具有优良药用性状的品种奠定了基础。
利益冲突所有作者均声明不存在利益冲突
参考文献(略)
来 源:毛常清,沙秀芬,黄 静,陶 珊,彭 芳,李 群,张 超,袁 灿.川芎基因组survey测序及其特征分析 [J]. 中草药, 2023, 54(3):907-914.
- 本文固定链接: https://maimengkong.com/zixun/1367.html
- 转载请注明: : 萌小白 2023年1月29日 于 卖萌控的博客 发表
- 百度已收录
