细胞器基因组研究相对于微生物基因组来说,要相对容易的多,因为细胞器基因组不大(高等植物叶绿体基因组大小基本在130~160kb左右,动物线粒体基因组基本都在20kb以内),拷贝数高,GC含量与基因组有较大差别(针对大部分物种),所以在高通量测序技术应用非常普通的今天,细胞器基因组很容易从total DNA测序数据中拼接分离出来。而且,细胞器基因组的基因构成相对很稳定,所以细胞器基因组研究的初步工作,如拼接注释,大家通过读读文献或者百度google就能够学会或者解决。
当然,我今天要谈的核心不是这些。虽然细胞器基因组的拼接和测序仍然有很多问题(物种本身或者测序污染啊等等导致拼接的困难啊之类的,总之问题很多),但是大部分实验室基本都不会卡在测序上,而是卡在分析上。
细胞器基因组分析里边包括很多的分析点,但基因分析都涵盖了结构比较、基因内容比较、基因组线性分析和系统进化分析等等。噢,今天谈的就是这个系统进化分析。

有人会说,系统进化分析有啥难的,不就建颗树么,比对比对,扔进软件里,谁还不会?(对,我们就是坚实的MEGA党)
嗯~,这么说也没问题,大部分人都可能建过树,因为本身构建进化树就跟大象装冰箱一样分为三个步骤:序列收集,序列比对,和扔进软件建树。
不过,讲真,我可真不能讲系统进化分析就很容易,因为被虐过进化分析的我,深知系统进化的可怕,在高通量测序还未普及的以前,系统进化占据着生信分析的大山头。我突然又想起被系统进化支配过的恐惧(理论超级不友好,非常虐,大部分系统进化的软件也很不友好,都需要看大量的手册和指导文章)。
我们继续回到正题,嗯~,细胞器基因组。
系统进化分析基本是细胞器基因组文章的“家常菜”,就是利用细胞器基因组构建系统发生树,解决或者验证一些物种进化亲缘关系的问题。
通常,去做这样的分析,在细胞器基因组研究里,是需要构建多基因比对序列的,也就是把所有涉及的基因组中的直系同源基因都预先抽取取出来,进行单独比对(当然这个比对过程也有很多道道),然后把比对好的每一个基因在按照物种合并起来,构建出含有全部或者大部分基因组的、基因组层面的比对序列。然后再用这个去建树。
OK,假设,你是一个很好的生信winner,针对这个步骤,你也得写个流程,把那些基因需要从基因组中抽出来,然后进行合并,然后让他们自动比对......我貌似记得有个这么个流程,可以把直接同源基因都凑在一块,不过仍然是手动得精力耗费要大于自动化流程。
OK,也不用假设,你没啥生信基础,这个工作怎么做?可能会利用下gb注释文件,在各种综合软件里,如Ugene,Genious里手动挑出那些编码基因序列,然后一个基因黏在同一个文本文档里。好不容易一个基因粘贴完了,下边进行比对了,你会有时候发现,这个基因怎么少了这么大一块,跟别的序列长度差太大啊,哪里出问题了?然后回到源头去找。这个工作就反反复复,没完没了。
OK,假设你很有野心,我要建个大树,物种多多。嗯,比方说有100个物种吧,按照动物线粒体13个蛋白编码基因吧,还不考虑非编码基因,那么光抽序列,我得抽1300条,还不能出错(出错就得重来),我算算啊,日抽序列200条,嗯,得花一个周来抽序列,然后下个周把他们比对,然后在下个周把序列合并。我咧个去,如果合并过程发现了问题(比如有几条序列总长度不一致啦),回头看那一堆茫茫序列,根本不值到从何找起。
大问一声怎么办?重做吧。
笔者,就是这样,就是为了搞个树,我能晃晃悠悠花一个月,有木有!(想起来就是一把辛酸泪)
其实我也很同情那些搞叶绿体基因组的,因为叶绿体基因组往往要用到几十个基因,通常叶绿体基因组共有的蛋白编码基因有76个。所以搞叶绿体基因组树的同学,唉,好比要处理30个基因组,76X30=2280!呵呵呵~。
我不知道有多少搞细胞器基因组的童鞋,不知道他们的痛苦是不是跟我一样。
今天就是要介绍下我的解决方案,准确的说是介绍一个流程:HomBlocks。
先说一下,这个HomBlocks的功能和优点。
HomBlocks是雷神之锤写的一个很简单的perl脚本,它能够完成上述的工作,给它100个动物线粒体,它能半个小时之内就能给你直接用来建树的序列。
它的优点就是快,就是高效。半个月,一个月才能搞定的建树比对序列,HomBlocks不到一天就能完成(最长花了大概8个小时,构建70多个叶绿体的比对序列),而且质量高。
以下是HomBlocks的工作原理:
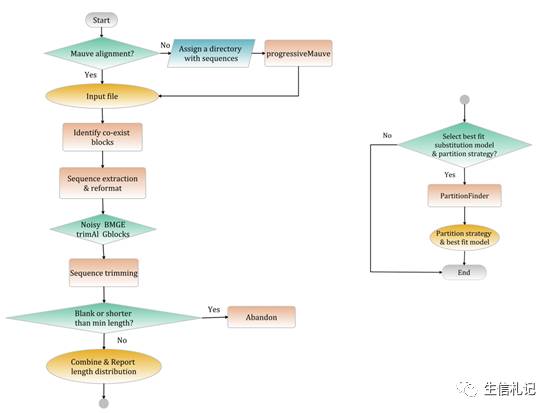
可能眼尖的童鞋一下就看到了, 哦,原来你是用了mauve啊,这个软件我知道,用来做基因组共线性分析的啊,怎么跟细胞器基因组序列比对有关系呢?你用它,我不放心哦。
在解释这个问题之前,其实还有个问题需要提一下,但实质是一样的,很多人会问:你要建细胞器基因组的系统进化分析,直接用基因组序列去比对不就好了?这个方法岂不是更高效?
唔~,其实并不是这样。首先,很多序列,起始位置并不一致,虽然细胞器基因组大都是环形的,可是序列起始位置都是个人自行决定的,你要进行基因组直接比对,你得调整序列位置吧,这个事不比上边讲的那个事容易。其次,很多基因组并不保守,序列中有插入啊,长短不一致啊,内含子多样啊,结构变化啊都是很常见的现象。诺~,如下图所示。
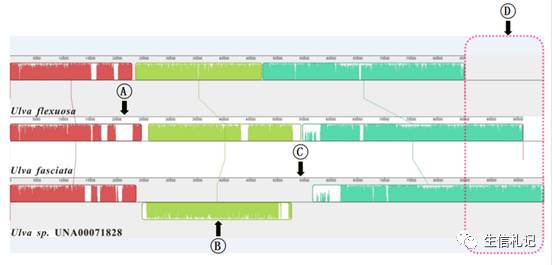
所以,细胞器基因组无法直接比对哦~。
即便序列长度差不多,而且很保守,如果直接扔进比对软件,比如mafft,时间也会很长的哦~,甚至根本跑不动。
再来回答刚才的问题,为什么用mauve。
Mauve这个软件非常牛逼,这是迄今为止唯二使用比对锚定算法的软件之一。怎么讲呢,通俗的讲,就是可以通过基因组序列直接来搜寻基因组共线性模块(locally collinear blocks)。这样我们通过mauve,就把全基因组的基因序列比对问题转换为全基因组共有的线性模块搜索问题。
也就是,把基因比对,变成了广义的模块比对。
嗯~,然后搜索所有基因组共存在的模块,就可以拿来建树啦,当然这些模块会经过序列比对修饰软件的处理(如Gblocks等)。
不知道童鞋们听懂了没。
HomBlocks除了高效,而且不需要基因组注释,单纯的fasta序列就好,是不是非常简单呐,当你还在辛辛苦苦的注释时候,就已经可以建树了哦。
有童鞋会问HomBlocks除了速度快,真正的效果怎么样?
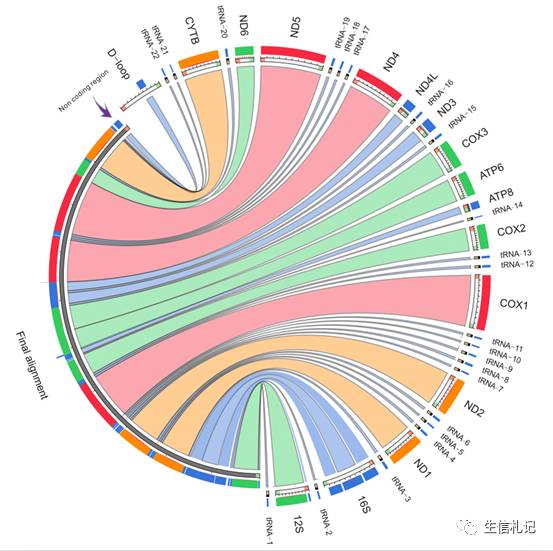
其实HomBlocks自带了两个例子,一个是36啮齿动物线粒体的的比对,这个可以在原网址上看到,比对过程大概10分钟不到吧。
我们看下这个circos图,看下效果,左边是最终生成的比对序列(长度15170bp),左边是对应的基因,显示了这些基因的收容情况和相对位置(其实就是个blast图啦)。可见HomBlocks构建的alignment几乎包括了所有的基因,然后还有个D-loop区的一个保守的片段。是不是很全面啊~。
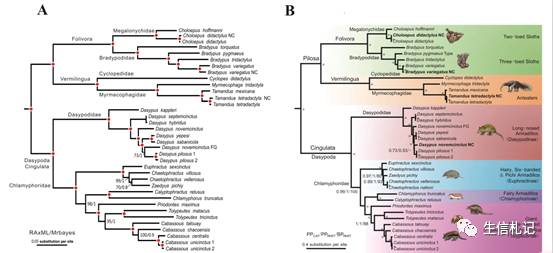
它这36个物种使用的数据和一篇MBE文章的数据是一样的,用HomBlocks构建的alignment进行建树,和原文进行下比较。
当当,图A自然是自己建的树啦,图B是MBE原文的树,怎么样,除了枝的长短可能有差别,树形很一致吧。
除了线粒体,叶绿体基因组的例子也有。来来猜猜哪个是用HomBlocks alignment建的树,哪个是原文的。
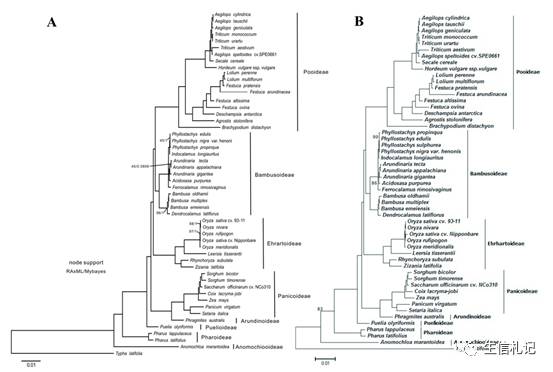
所以,利用HomBlocks你完全可以一天之内get到你想要的树~。不用在被老板催啦~
HomBlocks下载地址:
详细教程还是参看 readme。
一天之内拿到树,你也可以!- 本文固定链接: https://maimengkong.com/kyjc/1413.html
- 转载请注明: : 萌小白 2023年3月29日 于 卖萌控的博客 发表
- 百度已收录
