2024
09-29
09-29
干货|除了SCI,还有哪些期刊评价指标?
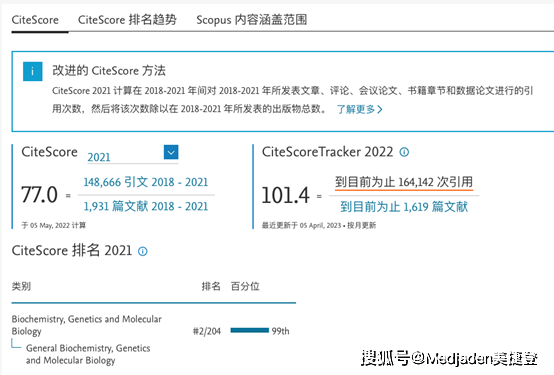 目前国际上最常用的期刊评价指标就是SCI。SCI的评级指标已经对科学发展产生了巨大影响,但也暴露出一些缺陷:如易被人为操纵、统计错误、不能跨学科比较、选源标准问题,对非英文期刊不公平,以及假设“所有引文重要性等价”等。影响因子对于期刊的评价并不是非常完美的。所以,今年来,越来越多的期刊评价标准被开发。那么,除了SCI之外,还有哪些常见的期刊评价指标呢?一、谷歌学术H5指数由谷歌学术...阅读全文&... 阅 读 全 部 >
目前国际上最常用的期刊评价指标就是SCI。SCI的评级指标已经对科学发展产生了巨大影响,但也暴露出一些缺陷:如易被人为操纵、统计错误、不能跨学科比较、选源标准问题,对非英文期刊不公平,以及假设“所有引文重要性等价”等。影响因子对于期刊的评价并不是非常完美的。所以,今年来,越来越多的期刊评价标准被开发。那么,除了SCI之外,还有哪些常见的期刊评价指标呢?一、谷歌学术H5指数由谷歌学术...阅读全文&... 阅 读 全 部 >
 SCI期刊发表之前必须查重吗? Sci期刊投稿前要不要查重,有的作者想要发表sci论文,但是不知道发表之前是不是必须要查。重,今天我们就来说一下这个问题。 Sci期刊发表之前是必须要查重的。一般来说要求不能高于20%,也有一些期刊要求不能高于15%,对于一些要求最为苛刻的期刊来说是要求不能超过5%的,不过这种期刊相对来说数量比较少,而且大多是一些顶刊或者是高分期刊。 那么有的作者会觉得我投稿之前要...
SCI期刊发表之前必须查重吗? Sci期刊投稿前要不要查重,有的作者想要发表sci论文,但是不知道发表之前是不是必须要查。重,今天我们就来说一下这个问题。 Sci期刊发表之前是必须要查重的。一般来说要求不能高于20%,也有一些期刊要求不能高于15%,对于一些要求最为苛刻的期刊来说是要求不能超过5%的,不过这种期刊相对来说数量比较少,而且大多是一些顶刊或者是高分期刊。 那么有的作者会觉得我投稿之前要...  大多数SCI期刊都采用在线投稿系统,文章投稿成功后,你就可以通过投稿系统查看文章的状态。今天我们就来说说投稿系统中各个状态的意思。1. 系统显示"Submitted to journal"——提示文章投递成功,同时通讯作者的邮箱也会收到一封Confirmation Email。在文章最初投递出去的这几天,要密切关注通讯作者邮箱和投稿系统,因为文章很可能因为文章格式不符...阅读全文>>...
大多数SCI期刊都采用在线投稿系统,文章投稿成功后,你就可以通过投稿系统查看文章的状态。今天我们就来说说投稿系统中各个状态的意思。1. 系统显示"Submitted to journal"——提示文章投递成功,同时通讯作者的邮箱也会收到一封Confirmation Email。在文章最初投递出去的这几天,要密切关注通讯作者邮箱和投稿系统,因为文章很可能因为文章格式不符...阅读全文>>...  蛋白激酶是一类在细胞内发挥关键调控作用的蛋白质,被广泛视为细胞调控网络的“主宰者”,其在细胞信号传导、生理调节和疾病治疗领域中的关键作用,使其荣膺生物医学研究中的“重要之星”,是药物研发中备受关注的靶点之一。2022年,FDA批准上市的37种新药中,就有5种药物属于小分子激酶调节剂。针对激酶的药物研究旨在调控相关生物学过程,从而对抗疾病。作为生物分子世界中的调控“大师级”元素,如何应用生信分析解析...
蛋白激酶是一类在细胞内发挥关键调控作用的蛋白质,被广泛视为细胞调控网络的“主宰者”,其在细胞信号传导、生理调节和疾病治疗领域中的关键作用,使其荣膺生物医学研究中的“重要之星”,是药物研发中备受关注的靶点之一。2022年,FDA批准上市的37种新药中,就有5种药物属于小分子激酶调节剂。针对激酶的药物研究旨在调控相关生物学过程,从而对抗疾病。作为生物分子世界中的调控“大师级”元素,如何应用生信分析解析... 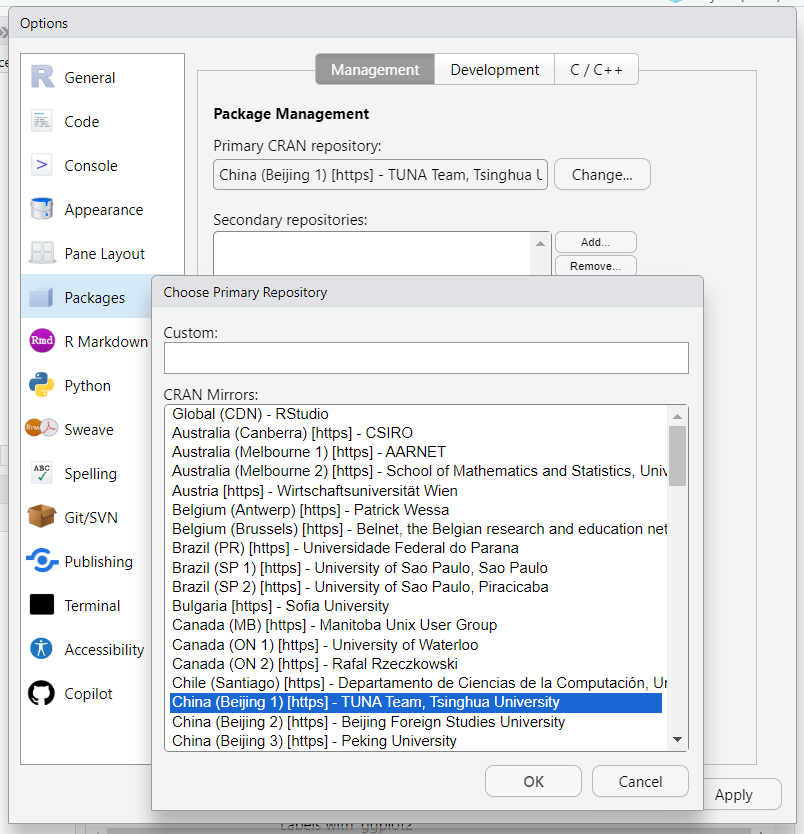 CRAN安装R包CRAN (Comprehensive R Archive Network) 是 R 语言的官方仓库,几乎所有主流的 R 包都可以通过 CRAN 安装。镜像设置建议使用CRAN安装R包前,将镜像设置为离您较近的镜像点,这里推荐清华大学提供的CRAN镜像,选择后,保存即可。CRAN镜像设置安装时可以点击Packages标签内的Instal...阅读全文>>...
CRAN安装R包CRAN (Comprehensive R Archive Network) 是 R 语言的官方仓库,几乎所有主流的 R 包都可以通过 CRAN 安装。镜像设置建议使用CRAN安装R包前,将镜像设置为离您较近的镜像点,这里推荐清华大学提供的CRAN镜像,选择后,保存即可。CRAN镜像设置安装时可以点击Packages标签内的Instal...阅读全文>>... 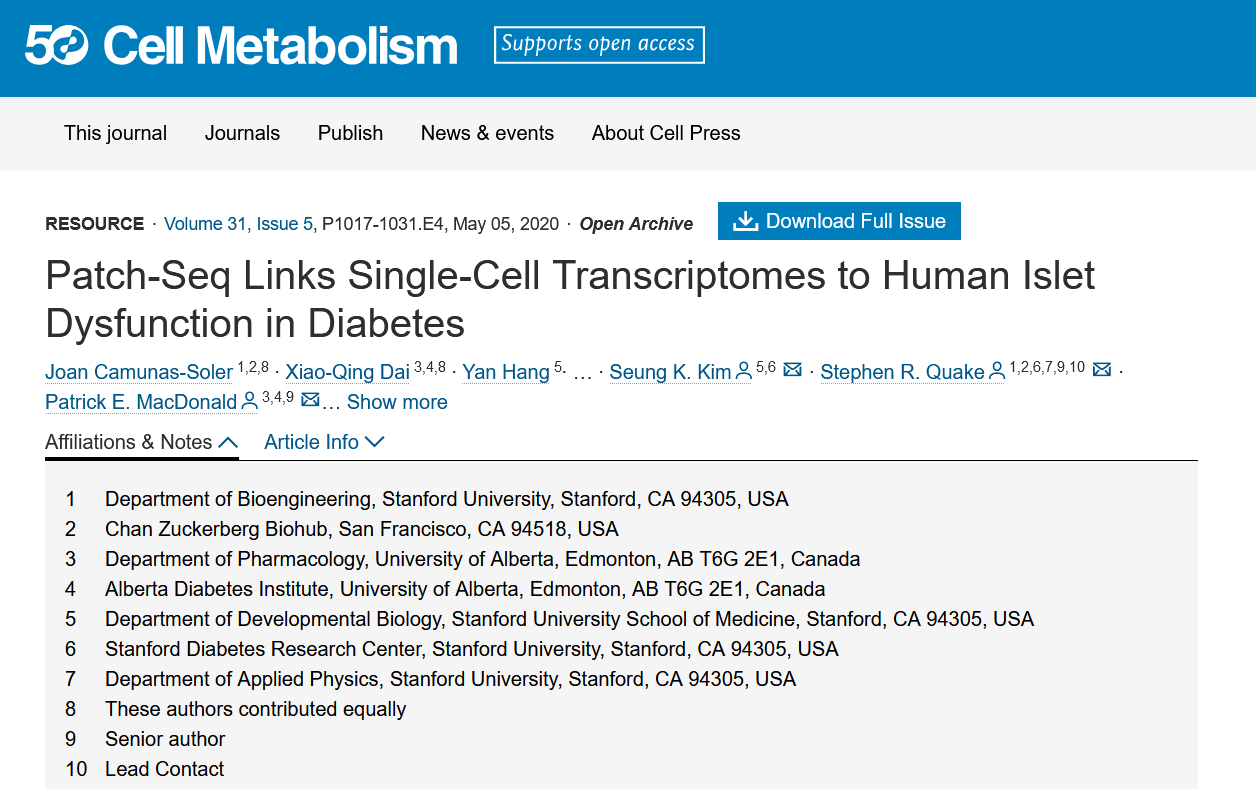 Patch-seq是一种结合了膜片钳记录技术(patch-clamp electrophysiology)、单细胞转录组测序(single-cell RNA sequencing)和形态学分析的新兴技术。这种方法能够在同一个细胞中同时获得电生理特性、基因表达数据和形态学特征。2020年在细胞代谢期刊上发表了题为《Patch-Seq Links Single-Cell T...阅读全文>&...
Patch-seq是一种结合了膜片钳记录技术(patch-clamp electrophysiology)、单细胞转录组测序(single-cell RNA sequencing)和形态学分析的新兴技术。这种方法能够在同一个细胞中同时获得电生理特性、基因表达数据和形态学特征。2020年在细胞代谢期刊上发表了题为《Patch-Seq Links Single-Cell T...阅读全文>&... 