高通量测序时代,大量的原始测序数据都流向了NCBI的SRA数据库。尽心尽责的将文章有关的测序数据上传至公共平台发布,一方面,是对生物领域科学研究的奉献,方便数据再利用,进行新探索;另一方面,可进行数据分析重现、验证等,为课题研究的真实性提供依据。
但是,很多操作过数据上传的小伙伴们都发现,认真做好这件事可真不容易,成功拿到传说中的SRA accession number(序列检索号码)仿佛经历了九九八十一难。从申请开始,就会有各种莫名的“error”、“warning”从天而降,好不容易走到最后一步,可能数据上传又出错了,或者怎么也传不上去……
今天我汇总自己的上传经历以及被频繁咨询的各种上传问题,给大家分享整理汇总好的一套操作指南,内含丰富的友情提示,防报错防警告你值得拥有!
SRA数据库简介
数据库地址:
https://www.ncbi.nlm.nih.gov/sra
SRA(Sequence Read Archieve)数据库是由美国国家生物技术信息中心 (National Center of Biotechnology Information)搭建的存放原始测序数据的地方。SRA兼容不同测序平台的数据,比如Sanger测序、Illumina测序等,所以无论是转录组、微生物扩增子,还是宏基因组等组学分析,都可以将原始测序数据上传SRA。SRA是使用率最为广泛的数据上传平台。此外,SRA与EBI(European Bioinformatics Institute)、GEO(Gene Expression Omnibus)等其他数据库都存在数据关联共享。所以SRA是大部分科研小伙伴分享测序数据的首选。
文献使用示例
数据上传完成后会得到非常多的ID。我们挑选可用的ID后,一般在文献的材料方法部分指明将原始测序数据(raw sequence data)上传在了NCBI的SRA数据库(图1)。那么有需要的小伙伴就可以在SRA库中搜索ID,精准查到相关数据。

图1 文献描述示例
数据上传
Tips 数据上传会遇到一些问题,如果自己解决不了,可以直接跟SRA后台发邮件沟通,他们的处理效率非常高而且非常耐心,一般2天左右就会回复邮件。
针对SRA上传相关问题的联系邮箱:sra@ncbi.nlm.nih.gov
数据上传地址:https://submit.ncbi.nlm.nih.gov/subs/sra/
在SRA平台注册账号登录后,即可创建上传任务。总体有8个步骤(图2)。主要包含相关信息填写(1至6步)和测序数据上传(0或7步)。
Tips账号注册申请后需要进注册时填写的邮箱验证通过,否则只能登录,界面没有“New submission”的选项。

图2 数据上传步骤总览
1.Submitter
登录后,界面中有“New submission”的选项(图3),点击即可创建上传任务。
Tips未完成的上传任务,SRA会保存已填写的结果,所以支持不连续填写。下次填写,可在界面底部的任务记录,直接点击任务编号,进入未完成的任务。
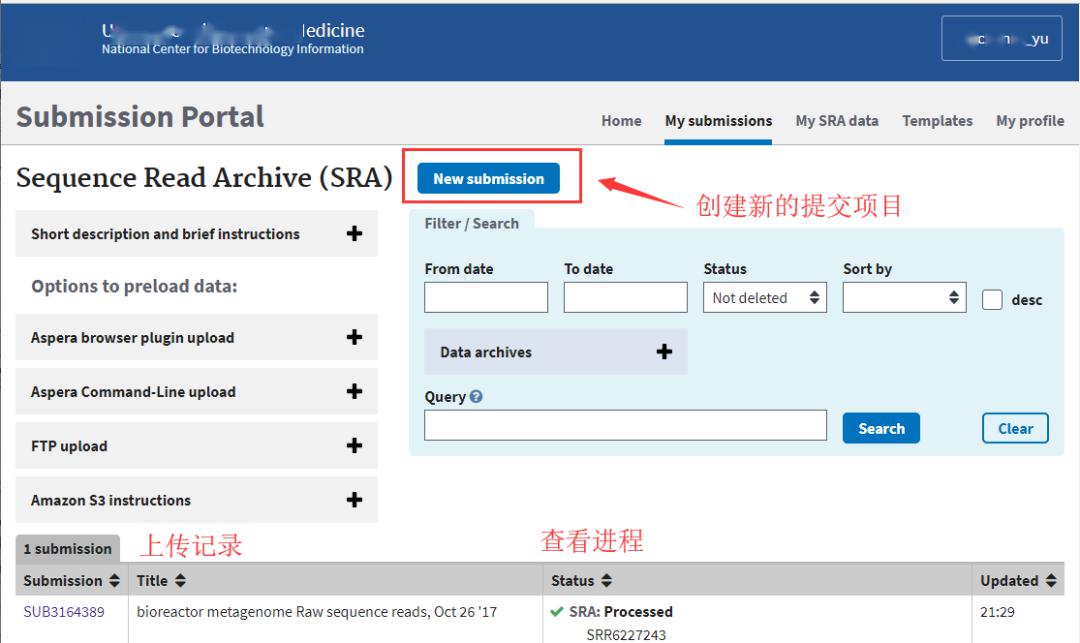
图3 New submission界面
点击创建任务后,即进入上传者信息(submitter)界面(图4),此时会获得一个以SUB开头的编号,用于记录上传进程。在该界面填写完上传人姓名、邮箱信息,点击“continue”。
Tips 星号“ * ”为必填项,其他可不填。由于 qq.com,163.com, foxmail.com 可能拒收NCBI邮件,为确保接收需填写这三类邮箱以外的备用(secondary)邮箱,如学校、单位邮箱等,否则点击“continue”后界面无法跳转。

图4 Submitter界面
2.问卷(General)
跳转至General界面(图5),调查3个问题:
1)是否创建过Bioproject(研究课题描述);
2)是否创建过Biosample(样本信息描述,如样本类型、采样点信息等);
3)选择数据发布时间。
因为有些大型项目,可能多年采集样本分批测序,所以会共用一个Bioproject;或者同一份样本同时进行了16S和ITS测序,则可以共用一个Biosample。所以NCBI支持调用以前的信息,避免重复填写。
在该界面根据自己数据的情况,若已有Bioproject和Biosample,则勾选“Yes”,并填写PRJNA开头的Bioproject编号,方便SRA进行匹配;若没有,则勾选“No”。
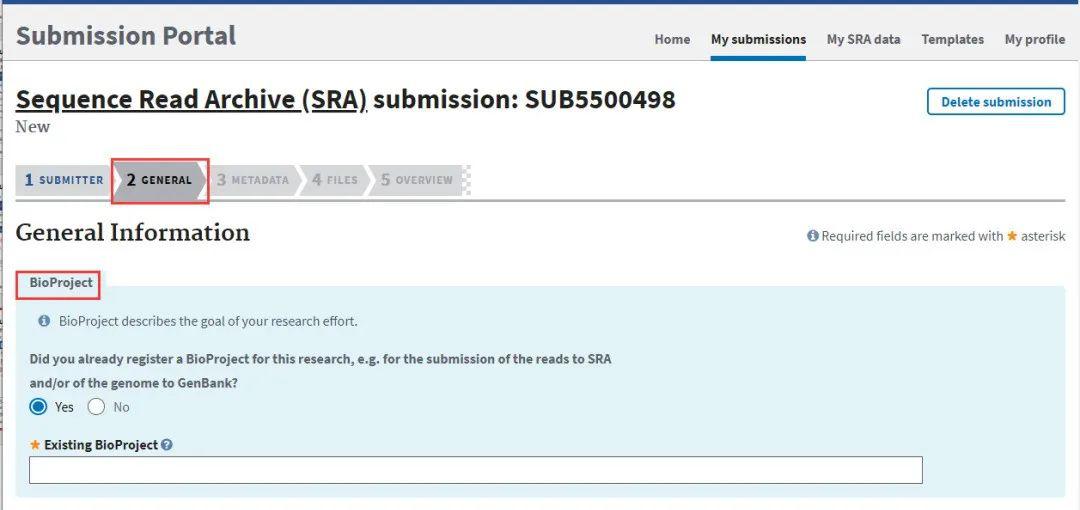
图5-1 Genera-Bioproject界面
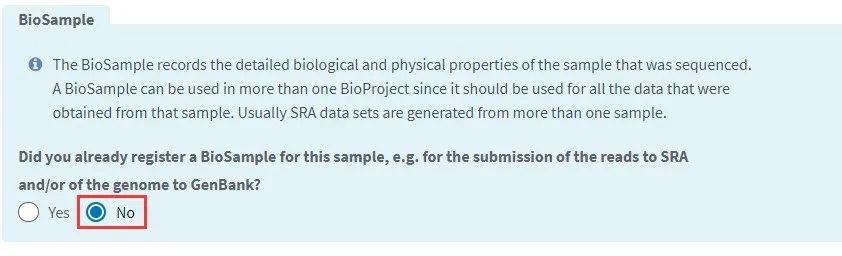
图5-2 Genera-Biosample界面
然后是数据发布时间选择(图5-3)。数据状态分上传和发布两种,完成上传后,表示数据已存放至SRA平台,但是其他人通过关键词搜索等无法查到这部分数据;完成发布,即Release,则其他人检索可以查到该数据。由于一般情况都是先上传数据,但文章还没正式发表,所以不想立即公开测序数据。
SRA提供了2个选择:
1)上传完成后立即发布;
2)自定义发布时间,默认只能选择一年以内的时间范围,即最晚也需要在一年后释放。
推荐自定义。其他信息选填,可结合项目信息继续完善。完成后点击“continue”。
Tips如果需要更改发布时间或者临时查看,比如遇到数据还未发布但审稿人需要查看,有两种处理方式:1)在登录界面的“Manage data”栏目点击“create reviewer link”生成一个数据浏览的链接,即检索不到但可以进入链接查看;2)给SRA后台发邮件请求更改发布时间。

图5-3 Genera-Release data界面
3.Project info
跳转至Project info界面(图6),填写数据对应的研究课题,项目标题、内容简介等,方便查询数据的人了解相关背景。内容简介不限定字数,详略都可,也可引用文章摘要。

图6 Project info界面
4.Biosample type
跳转至样本类型选择(图7-1),根据上传数据的样本特征勾选。SRA提供了10种样本类型,比如RNA-seq可能对应“Microbe”(单菌株)、“Model organism or animal”(经典的模式物种)、“Human”(人)、“Plant”(植物)。
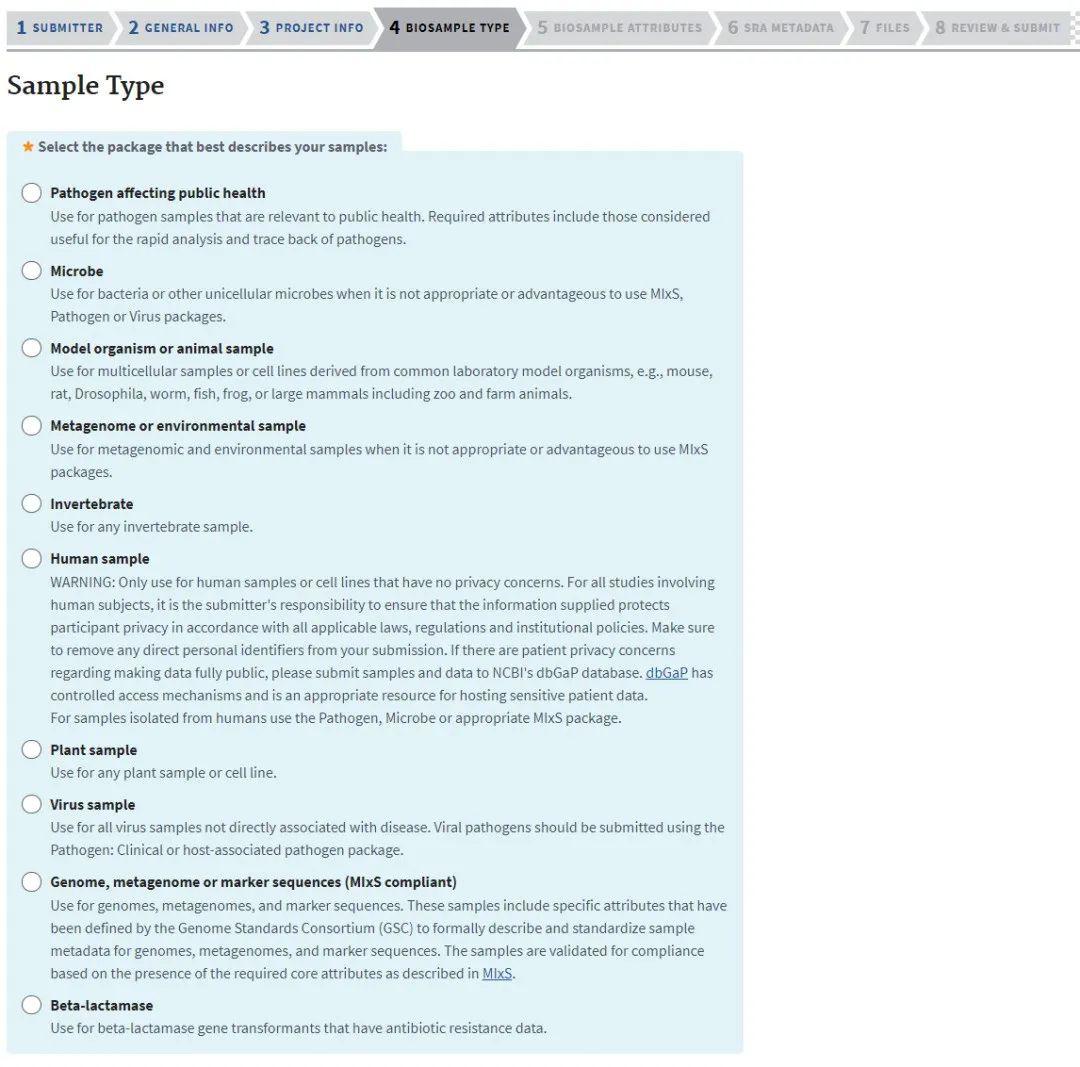
图7-1 Biosample type界面
对于微生物群落的扩增子、宏基因组等测序分析,可能对应“Genome, metagenome or marker sequences (MIxS compliant)”(基因组序列分析)或者“Metagenome or environmental”(宏样本或环境样本)。
具体的,“Genome, metagenome or marker sequences (MIxS compliant)”包含9种类型(图7-2),一般对应“Environmental/Metagenome Genomic Sequences (MIMS)”,勾选后又有15种类型(图7-3),比如空气、宿主相关的、沉积物、土壤、废水、自然环境水体等,可根据样本特征勾选。如果以上15种都不符合,比如青贮发酵样本,则选择“Metagenome or environmental sample”。
下一步,针对选择的样本类型,会有对应的样本属性表格,所以如果样本类型不合适,会影响后续属性填写。
Tips每种样本属性表格都有必填列,如果属性类别不符合样本特征,比如你本来是环境样本,遇到有宿主信息必填,说明样本类型不合适,可回到该步骤重新选择。

图7-2 基因组分析样本类型

图7-3群落分析样本类型
5.Attributes
跳转至“Attributes”样本属性填写的界面(图8-1)。支持网页端填写和Excel模板导出本地填写。样本少可以网页输入;样本很多,以防断网中断等,建议导出模板。
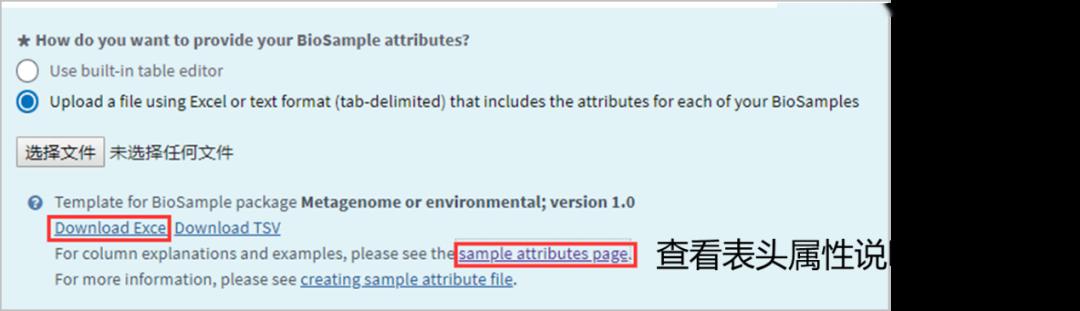
图8-1 Biosample attribute 界面
以“Metagenome or environmental sample”类型的本地excel为例(图8-2,8-3),绿色为必填列,蓝色为至少填其中一列,黄色为选填列。一个样本一行,需要按照表头的帮助文档规范填写,如采样点填写,国家的城市,必须以冒号分隔(图8-2)。黄色列多与实验设计相关,可以尽量准确描述。表格末尾可以自定义输入表头,添加列信息。填写完成后上传该文档,跳转至下一步。
Tips不一定所有实验都采集了经纬度信息,如果没有可填写“not collected”,且不可以是其他字符,否则SRA不识别,无法下一步。
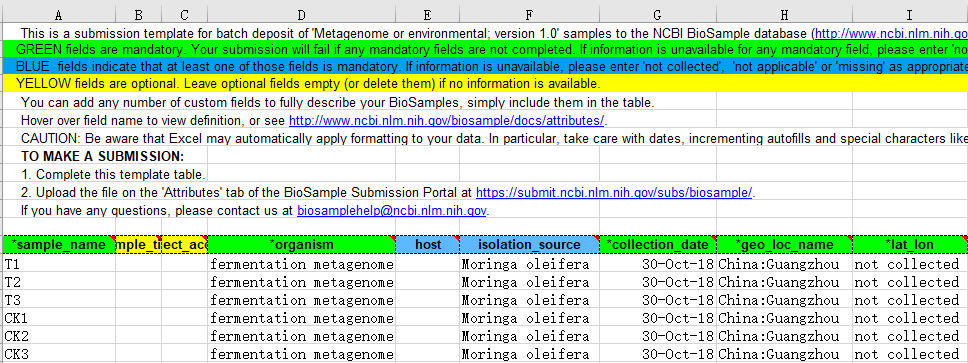
图8-2 样本属性表格(一)
Tips除了样本名/标题,行与行之间的描述信息(其他列)不能完全一样,否则会报错,无法跳转下一步。如,某两行可以有5列填写完全一样,但有1列填写不一样。一般分组之间,可能因为采样点不同或者实验处理条件不同,所以不会重复。但是同一个分组内的生物学重复样本,所有列都一样,这是最频繁的报错之一。解决方法,如图8-3,在末尾添加一列“replicate”,对组内的重复样本标记序号,即可避免组内样本描述重复。
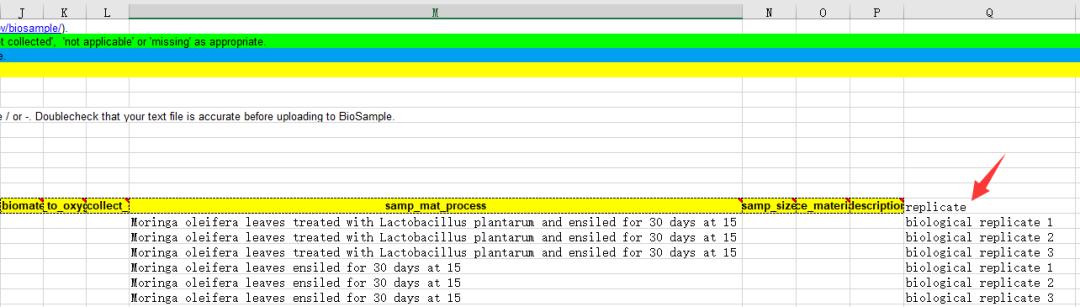
图8-3 样本属性表格(二)
6.Metadata
如图9-1,用于SRA后台匹配样本属性描述和样本测序信息。
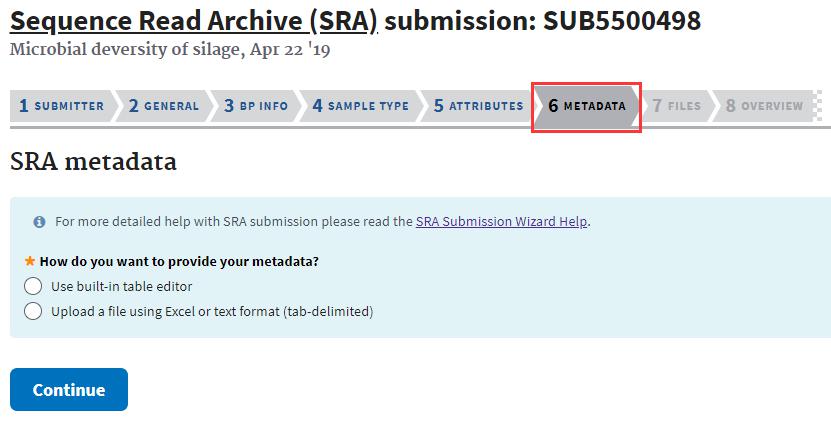
图9-1 Metadata界面
以导出模板的本地填写为例。除第一列以外,蓝色为必填列,其余为选填。16S等扩增子测序,则“Library_strategy”选择“AMPLICON”,“library source”选择“METAGENOMIC”,“library selection”选择“PCR”。若为宏基因组测序,则依次选 “WGS”、“METAGENOMIC”、“RANDOM”。其他列则根据测序平台对应选择(图9-2)。

图9-2 metadata表格(一)
然后是实验描述、序列类型、文档名(图9-3),Illumina平台测序数据格式一般都为fastq格式。filename列十分重要,因为SRA的后台会基于文档名对应匹配上传的数据,也是大家遇到报错最多的一步。
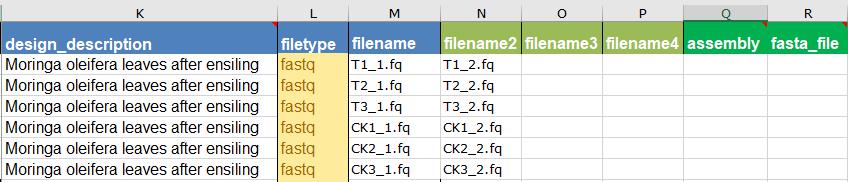
图9-3 metadata表格(二)
Tips一个样本的测序数据放一行,如双端测序,或者因序列文件太大而拆成了2个文档,这种情况都写在同一行。一个文档写一列。
Tips表格的文档名与序列文件名必须完全一致。常见的书写错误,如表格填的名称出现了多余的空格,将“fq”写成了”fg”等等,所以如果第7步序列文件传完后,有报错说文档丢失,一定要回来metadata表格仔细核对检查文档名。
Tips 如果将10个序列文件压缩成一个文档上传,表格中不能写10个文档的压缩包名称,而应该写压缩包里每个序列的文档名。
填写完成后,即可进入第7步测序文件上传,下一期微信,我们将分享如何安装使用Aspera快速上传序列。- 本文固定链接: https://maimengkong.com/zu/1780.html
- 转载请注明: : 萌小白 2024年8月21日 于 卖萌控的博客 发表
- 百度已收录
