2024
02-06
02-06
采集卡软件之PotPlayer详情介绍
 采集并储存HDMI视频信号,不仅需要HDMI采集卡采集视频信号,也需要一些能够播放并储存视频信号的软件加以辅助。今天同三维就为您带来HDMI采集卡软件之PotPlayer详情介绍。首先,我们先来简单了解一下HDMI采集卡软件PotPlayer的由来:PotPlayer是由KMPlayer 的原作者使用 C++ 完全重写的播放器。它基本上继承了 KMPlayer 方...阅读全文>>... 阅 读 全 部 >
采集并储存HDMI视频信号,不仅需要HDMI采集卡采集视频信号,也需要一些能够播放并储存视频信号的软件加以辅助。今天同三维就为您带来HDMI采集卡软件之PotPlayer详情介绍。首先,我们先来简单了解一下HDMI采集卡软件PotPlayer的由来:PotPlayer是由KMPlayer 的原作者使用 C++ 完全重写的播放器。它基本上继承了 KMPlayer 方...阅读全文>>... 阅 读 全 部 >
 1.MusicBee这是一个免费的音乐管理器,可搜索本地硬盘、移动设备和互联网上的音乐文件,自动分类,我辛辛苦苦下载的周杰伦歌曲资源,总算在电脑上也能播放了。MusicBee 完全免费,可以帮我们创建多个音乐库,并且支持在多个音乐库间切换,支持 Boardcast 广播、视频播放、支持CD烧录、可由插件拓展更多功能。另外还可以快速搜索音乐等。2.CrystalDiskInfo...阅读全文>...
1.MusicBee这是一个免费的音乐管理器,可搜索本地硬盘、移动设备和互联网上的音乐文件,自动分类,我辛辛苦苦下载的周杰伦歌曲资源,总算在电脑上也能播放了。MusicBee 完全免费,可以帮我们创建多个音乐库,并且支持在多个音乐库间切换,支持 Boardcast 广播、视频播放、支持CD烧录、可由插件拓展更多功能。另外还可以快速搜索音乐等。2.CrystalDiskInfo...阅读全文>...  搞生物、基础医学同学如果对生命科学领域所有软件都了如指掌,那做事情必然会事半功倍。如果你对这些不了解,那就听师兄给你一一道来。下面的内容比较多,平时可以将下面的内容作为目录来收藏,权当做工具辞典来用。长话短说,切入正题。1. 三维分子类RASMOL:观看生物分子 3D 微观立体结构RasTop:为 RasMol 2.7.1 的图形用户界面软件...阅读全文>>...
搞生物、基础医学同学如果对生命科学领域所有软件都了如指掌,那做事情必然会事半功倍。如果你对这些不了解,那就听师兄给你一一道来。下面的内容比较多,平时可以将下面的内容作为目录来收藏,权当做工具辞典来用。长话短说,切入正题。1. 三维分子类RASMOL:观看生物分子 3D 微观立体结构RasTop:为 RasMol 2.7.1 的图形用户界面软件...阅读全文>>...  在科研领域“升级打怪”,怎能少了生物软件,以下是生物软件兵器库,任君挑选!1. 三维分子RASMOL:观看生物分子3D微观立体结构RasTop:为RasMol 2.7.1的图形用户界面软件CHIME:直接在浏览器中观看3D分子MolMol:将pdb等格式的蛋白文件通过微调,存成普通的图形文件raswin.exe.gz:rasmol(...阅读全文>>...
在科研领域“升级打怪”,怎能少了生物软件,以下是生物软件兵器库,任君挑选!1. 三维分子RASMOL:观看生物分子3D微观立体结构RasTop:为RasMol 2.7.1的图形用户界面软件CHIME:直接在浏览器中观看3D分子MolMol:将pdb等格式的蛋白文件通过微调,存成普通的图形文件raswin.exe.gz:rasmol(...阅读全文>>...  1、iLovePDFiLovePDF是一个非常强大的PDF处理和PDF转换网站,完全免费,功能丰富。包含丰富的PDF处理工具,比如合并PDF、拆分PDF、压缩PDF、PDF转Office、编辑PDF、PDF转图片、PDF加密和PDF解锁等等。iLovePDF目前有22个实用工具,界面简约,不需要注册登录就可以使用,操作简单,转换的效果也非常不错。2、智办事...阅读全文>>...
1、iLovePDFiLovePDF是一个非常强大的PDF处理和PDF转换网站,完全免费,功能丰富。包含丰富的PDF处理工具,比如合并PDF、拆分PDF、压缩PDF、PDF转Office、编辑PDF、PDF转图片、PDF加密和PDF解锁等等。iLovePDF目前有22个实用工具,界面简约,不需要注册登录就可以使用,操作简单,转换的效果也非常不错。2、智办事...阅读全文>>... 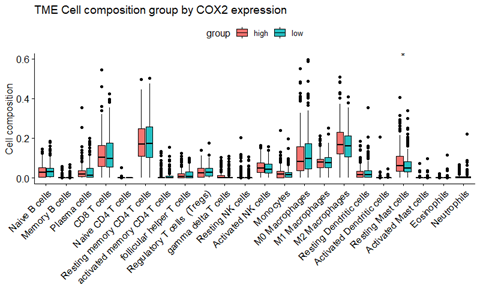 总结目前主流的免疫细胞浸润方法,整理归纳可用的资源及相关代码的实现,以及可视化方式,方便以后查询和分享。这里面提到的免疫浸润分析主要是基于组织样本转录组测序数据或者基因芯片数据的分析。因为我们取到的肿瘤组织并不只是包含肿瘤细胞,其中还有正常细胞、免疫细胞、基质细胞、血管细胞等。不同的细胞具有一些标志性的marker,免疫细胞也是一样。因此,可以根据这些marker基因在组织中的表达...阅读全文&...
总结目前主流的免疫细胞浸润方法,整理归纳可用的资源及相关代码的实现,以及可视化方式,方便以后查询和分享。这里面提到的免疫浸润分析主要是基于组织样本转录组测序数据或者基因芯片数据的分析。因为我们取到的肿瘤组织并不只是包含肿瘤细胞,其中还有正常细胞、免疫细胞、基质细胞、血管细胞等。不同的细胞具有一些标志性的marker,免疫细胞也是一样。因此,可以根据这些marker基因在组织中的表达...阅读全文&...  分享8个非常实用但是名气不大的软件1、uToolsuTools是一个非常强大的生产力工具箱软件,它自由集成了丰富的插件,可以快速匹配场景功能,用完即走。uTools优秀的插件化设计,让你可以自取所需,任意组合。插件一般仅数百KB大小,简洁美观、没有广告,解决你问题的,一个插件即可。uTools作者将此软件设计为”一切皆插件“的插件化工具,所有的功能均可通过插件实...阅读全文>>...
分享8个非常实用但是名气不大的软件1、uToolsuTools是一个非常强大的生产力工具箱软件,它自由集成了丰富的插件,可以快速匹配场景功能,用完即走。uTools优秀的插件化设计,让你可以自取所需,任意组合。插件一般仅数百KB大小,简洁美观、没有广告,解决你问题的,一个插件即可。uTools作者将此软件设计为”一切皆插件“的插件化工具,所有的功能均可通过插件实...阅读全文>>...  以下13款免费实用软件,全都是自用推荐,使用时长0.5-3年不等!第4款和第5款,简直不要太全能,一个抵千个!一、系统工具1、清理工具Glary Utilities一款简洁但强大的系统清理与优化工具,能够修复、增强、清理以及保护你的电脑。支持的功能有:一键清理电脑垃圾、注册表修复和清理、修复无效的快捷方式、重复/空文件查找、卸载电脑软件、进程管理、系统...阅读全文>>...
以下13款免费实用软件,全都是自用推荐,使用时长0.5-3年不等!第4款和第5款,简直不要太全能,一个抵千个!一、系统工具1、清理工具Glary Utilities一款简洁但强大的系统清理与优化工具,能够修复、增强、清理以及保护你的电脑。支持的功能有:一键清理电脑垃圾、注册表修复和清理、修复无效的快捷方式、重复/空文件查找、卸载电脑软件、进程管理、系统...阅读全文>>... 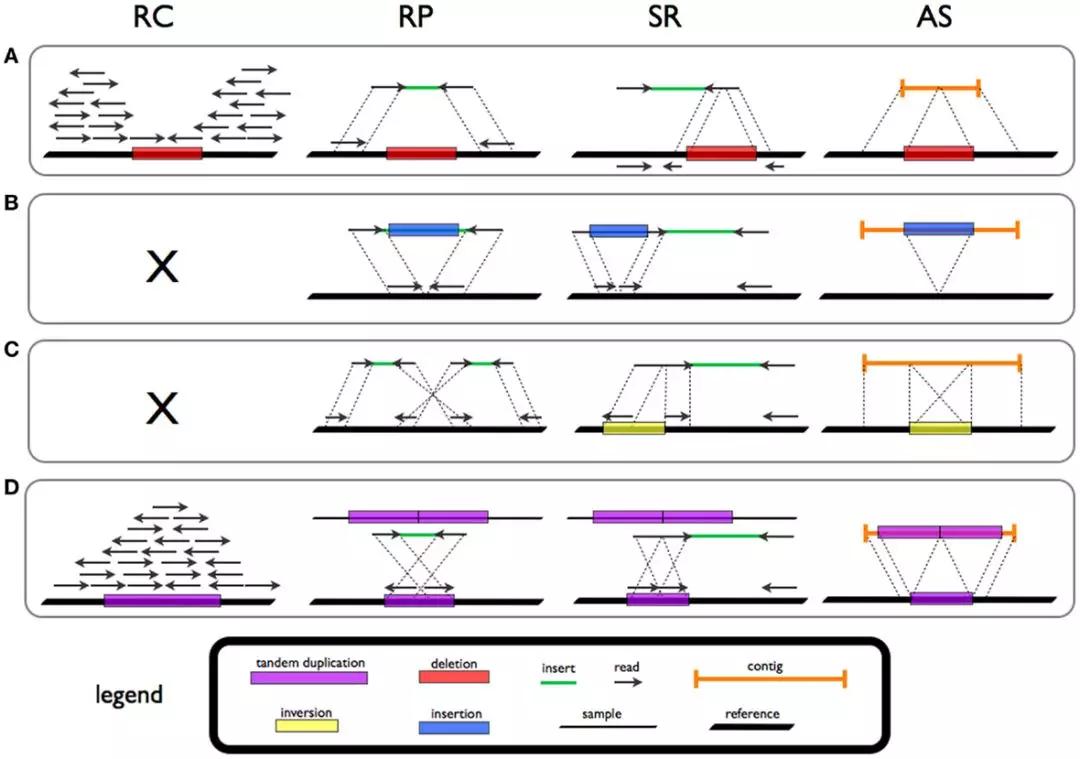 在 6 月份,基因组学领域的权威期刊Genome Biology发表了一篇方法学评估类文章,在这篇文章中作者系统地分类和评估了目前主要的69种基于全基因组测序(whole genome sequencing,WGS)数据分析结构变异的算法/方法:Comprehensive evaluation of structural variation detection algorithms for ......
在 6 月份,基因组学领域的权威期刊Genome Biology发表了一篇方法学评估类文章,在这篇文章中作者系统地分类和评估了目前主要的69种基于全基因组测序(whole genome sequencing,WGS)数据分析结构变异的算法/方法:Comprehensive evaluation of structural variation detection algorithms for ...... 