总结目前主流的免疫细胞浸润方法,整理归纳可用的资源及相关代码的实现,以及可视化方式,方便以后查询和分享。
这里面提到的免疫浸润分析主要是基于组织样本转录组测序数据或者基因芯片数据的分析。因为我们取到的肿瘤组织并不只是包含肿瘤细胞,其中还有正常细胞、免疫细胞、基质细胞、血管细胞等。不同的细胞具有一些标志性的marker,免疫细胞也是一样。因此,可以根据这些marker基因在组织中的表达量,结合一些生物信息学的算法,评估和量化免疫细胞浸润、免疫微环境。
对免疫微浸润的分析,本质上,就是搞清楚肿瘤组织当中免疫细胞的构成比例。对不同类型的肿瘤浸润免疫细胞进行定量研究,有助于阐明肿瘤免疫应答的机制、评价肿瘤治疗的免疫原性作用,最终指导设计合理治疗方案。当然,这个结果只是一个计算的推测结果,跟单细胞测序的实测相比更粗糙。优点是更经济实惠。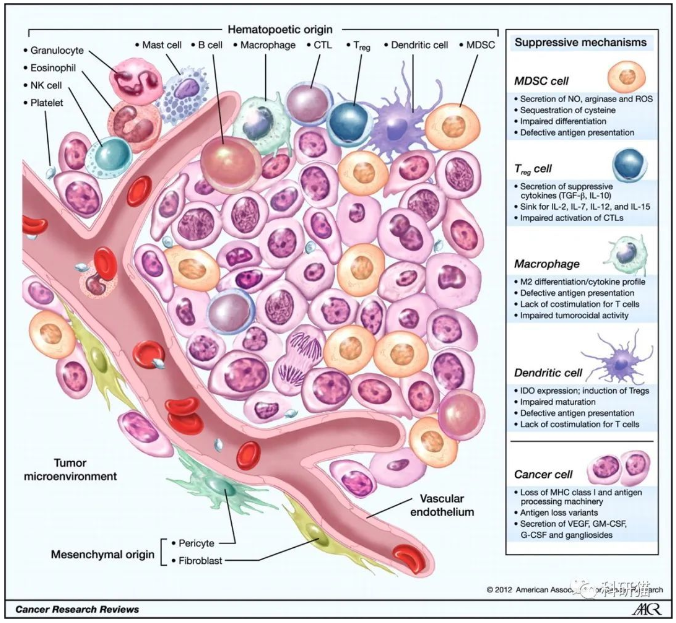
计算分析免疫浸润的方法主要有三类:基于cellMarker的,部分去卷积和完全去卷积的方法。


CIBERSORT
CIBERSORT是2015年发表于Nature Methods上的文章,是基于线性支持向量回归(linear support vector regression)的原理对人类免疫细胞亚型的表达矩阵进行去卷积的一个工具。该算法利用微阵列数据构建特征矩阵,描述22种免疫细胞表型的表达特征,包括不同的细胞类型和功能状态的免疫细胞。22种免疫细胞亚型的基因表达特征集:LM22(可以从网上下载)。
-
密码:XXXXX
-
这个方法推荐使用网页工具,因为操作简单快速。只需要准备整理好的测序数据集,首先上传,然后运行即可
- Signature Genes: LM22 CIBERSORT defau…
- Mixture file: HILC_TPM_data
结果解读
输出结果除了给出每个样本不同细胞的预测比例,后面还附带了三个统计量,p-value,R和RMSE。
-
CIBERSORT使用Monte Carlo采样对反卷积结果产生一个经验P值。这种方法首先假设给定的bulk RNA m GEP中不存在signature matrix(例如LM22)中的细胞类型,并使用皮尔逊相关分析计算m和f x B之间的统计量R。为了推导经验P值,CIBERSORT必须首先推导一个零分布R,因为与bulk RNA m GEP相比,签名矩阵signature matrix B只包含一小部分基因g。作者从bulk RNA m GEP中随机抽取g个基因表达值,形成一个随机的bulk RNA mI GEP,即|m| = |mi|;然后使用mi上运行CIBERSORT,生成估计细胞成分的向量fi。CIBERSORT然后计算随机bulk RNA mi与重组混合fi x B之间的相关系数Ri。此过程重复i次,得到R*。
-
P-value:在所有细胞子集上反褶积结果的统计显着性。
-
Pearson”s correlation coefficient (R):是通过将实际bulk RNA GEP与预测的bulk RNA GEP进行比较而得出的,后者是使用估算的细胞比例和来自签名矩阵signature matrix的相应表达谱进行计算出的。值得注意的是,这种相关性仅限于签名矩阵signature matrix标记基因。
-
RMSE:实际bulk RNA m GEP与预测的bulk RNA m GEP的均方根误差,仅限于签名矩阵signature matrix文件中的基因。
可视化分析
-
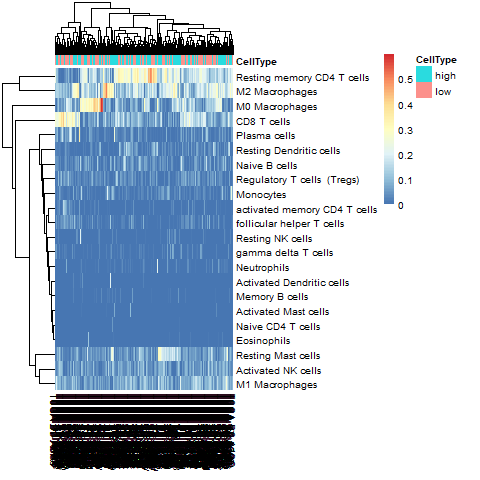
热图
 image-20210921162108457
image-20210921162108457 -
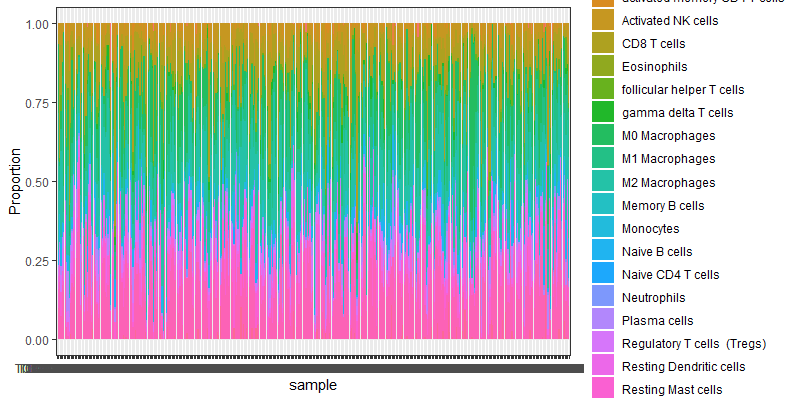
细胞比例图
-
 image-2021092116220620
image-2021092116220620 -
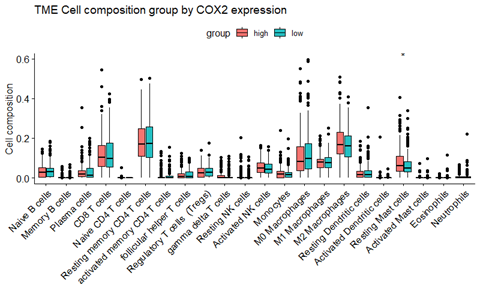
箱式图
-
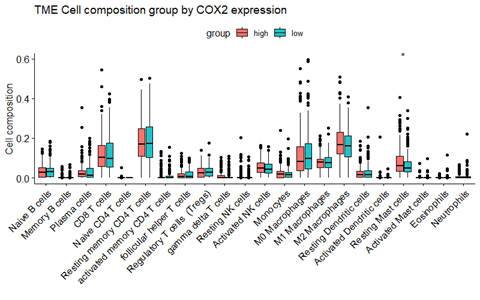 image-20210921162250619
image-20210921162250619
ssGSEA
单样本基因集富集分析(single sample gene set enrichment analysis, ssGSEA),是GSEA方法的扩展,计算每个样本和基因集配对的富集分数。每个ssGSEA富集评分代表了样本中特定基因集的成员被协调上调或下调的程度。相关文章于2009年发表于Nature:
-
Title: Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1。
-
Year: 2009
-
Journal: Nature
-
DOI: 10.1038/nature08460
GSEA生成一个基因集的富集得分,与一个数据集中的样本集合的表型差异有关。ssGSEA不同于以组别为单位(比如cancer vs normal)的GSEA分析,而是每个样本都可以得到相应基因集的评分。通过这种方式,ssGSEA将单个样本的基因表达谱转换为基因集富集谱。这种转化使研究人员能够根据生物过程和途径的活性水平而不是通过单个基因的表达水平来描述细胞状态。因此,ssGESA如果使用的是免疫细胞marker相关的基因集,就可以计算免疫细胞浸润评分。
算法原理
-
算法概括来说就是首先对给定样本的基因表达值进行秩次标准化,然后利用经验累积分布函数计算富集分数(ES)。
-
a. 首先假设有一个样本的表达数据,那么他应该是这样的,第一列为基因,第二列为表达值,这样的两列的数据矩阵。首先对样本的所有基因的表达水平进行排序获得其在所有基因中的秩次rank,这些基因的集合为BG。
-
b. 假设要对其进行KEGG的分析,首先我们需要在GSEA官网找到KEGG对应的gmt文件。gmt文件主要格式是:每行表示一个通路,第一列为通路ID,第二列为通路对应的描述,第三列开始到最后一列为该通路中的基因。
-
c. 那么对于任意的一个通路A,我们可以拿到这个通路的基因列表GL,从GL中寻找BG里存在的基因并计数为NC,并将这些基因的表达水平加和为SG。 开始计算ES:对于任意一个表达谱中的基因 G,如果G是集合GL中的基因则他的ES等于该基因的表达水平除以SG,否则记该基因的ES等于1除以(基因集合BG总个数减去NC)。 依次计算每个BG中的基因的ES值,找到其中绝对值最大的ES作为通路A的A.ES
-
d. 到此通路A的ES计算完毕,需要一个统计学方法来评估该ES是否是显著的,即非随机的。
-
按照上述计算ES的方法,先随机打乱表达谱中基因的表达顺序,然后再依次计算ES值,如此重复一千次,得到一千个ES值,我们根据这一千个ES值的分布,来计算A.ES在这个分布中所处的位置及出现在该位置时的概率即得到了p值 。依次分别计算每个通路的ES及p值,然后使用多重检验矫正得到每个通路的FDR。
实现方法
- 基因集需要是list为对象。
- 基因的表达量需要是矩阵,行为基因,列为样本。
默认情况下,kcdf=”Gaussian”,适用于输入表达式值连续的情况,如对数尺度的微阵列荧光单元、RNA-seq log-CPMs、log-RPKMs或log-TPMs。当输入表达式值是整数计数时,比如那些从RNA-seq实验中得到的值,那么这个参数应该设置为kcdf=”Poisson”。
具体的实现方法可以参考博客文章:http://www.biozhong.cn/categories/study/ssGSEA.html
TIMER
TIMER是一个用于系统分析不同类型癌症的免疫浸润。提供了多种免疫反褶积方法估计的免疫浸润丰度,允许用户动态生成高质量的数据,全面探索肿瘤的免疫学、临床和基因组特征。
可以利用一系列免疫特异性标记和免疫细胞表达特征,来估计32种癌症类型中6种免疫细胞类型(B细胞、CD4 T细胞、CD8 T细胞、中性粒细胞、巨噬细胞和树突状细胞)的丰度。
从RNA-seq或微阵列数据中提取的癌症表达矩阵与免疫细胞表达矩阵合并,并用Combat进行归一化,以消除批量效应。通过从免疫细胞标记中选择与肿瘤纯度负相关的基因,为每种癌症类型分别识别特征基因。最后,对于每种癌症类型,考虑到所选的免疫细胞标记,从标准化的免疫细胞配置文件构建特征矩阵。
TIMER使用线性最小二乘回归方法执行反褶积,并强制所有负估计为零。用越来越小的一组T细胞标记重复估计几次,以减少CD8 +和CD4 + T细胞比例之间的相关性。与CIBERSORT不同的是,最终的估计并没有被规范化,因此不能直接解释为细胞组分,也不能在不同的免疫细胞类型和数据集之间进行比较。
现在已经更新到TIMER2.0了
这个网站的资源很丰富,使用简单,集成了多种免疫浸润方法的数据,结合基因表达、突变、拷贝数变异、临床资料,可以做出很多漂亮的图。
功能
(1)可以计算基因、突变、ScNA以及临床结局与免疫细胞的相关性。
(2)如果是与临床结局相关,就可以得到生存分析的K-M图。
(3)两个基因在33中癌症中的相关性。
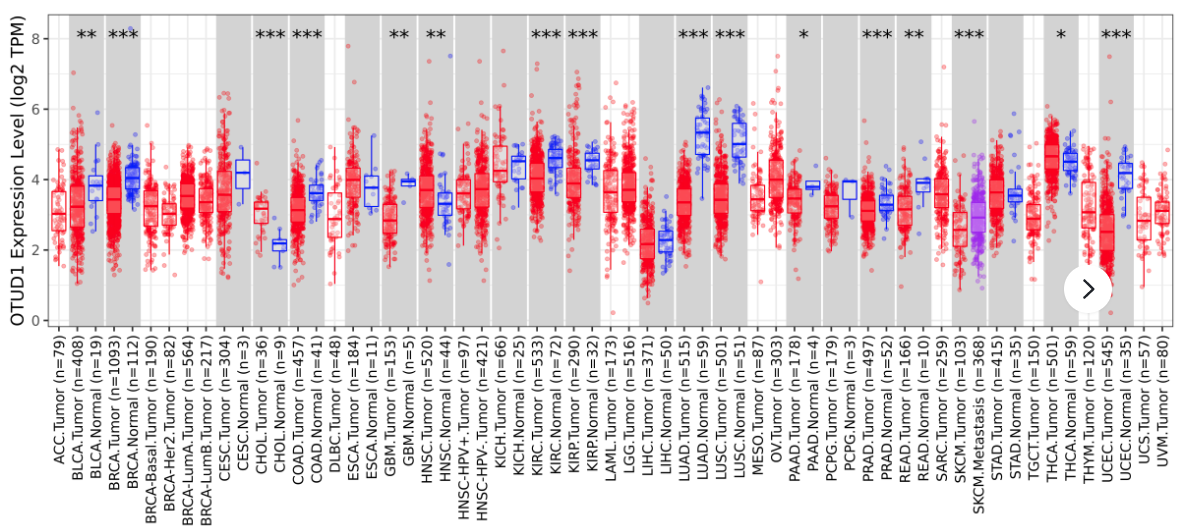
这里也可以计算一个基因与一组基因集中每个基因的相关性,比如免疫检查点的47个基因。
(4) 一个基因在33中癌症中的表达
(5)基因突变和基因表达以及基因突变与免疫细胞浸润的关系
MCP-counter
Microenvironment Cell Populations-counter,一种基于标记基因定量肿瘤浸润免疫细胞(CD3 + T细胞,CD8 + T细胞,细胞毒性淋巴细胞,NK细胞,B淋巴细胞,单核细胞来源的细胞(单核细胞系),髓样树突状细胞,中性粒细胞)、成纤维细胞和上皮细胞的方法。
对于每个细胞类型和样本,丰度得分为细胞类型特异性基因表达值的几何平均值,独立地对每个样本进行计算的。由于分数是用任意单位表示的,它们不能直接解释为细胞分数,也不能在细胞类型之间进行比较。进行定量验证时,估计分数与真实细胞分数之间具有很高的相关性,证明了MCP-counter用于样本间比较的价值。MCP-counter已应用于对32个非血液学肿瘤的19,000多个样本中的免疫细胞和非免疫细胞进行量化。
-
相关文章:Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression
-
R包:https://github.com/ebecht/MCPcounter
- 代码GZ04里面有
Xcell
xCell是一种基于ssGSEA的方法,能够计算64种免疫细胞的丰度分数,包括适应性和先天免疫细胞、造血祖细胞、上皮细胞和细胞外基质细胞。
作者整合了FANTOM、ENCODE、Blueprint、GEO数据库共1822个纯人类细胞型转录组,既包括RNA-seq也有Array-based数据,使用单样本GSEA(ssGSEA)分析方法对每个样本进行评分,获得每个样本最可靠的特征集(gene signature),共生成了489个特征。然后建模评估样本表达谱与特征之间的关联。最后作者将这个根据基因表达谱计算其各个细胞类型富集分数的工具。
xCell有网络工具,可以从64种免疫和基质细胞类型的基因表达数据中进行细胞类型富集分析。
也有R包:https://github.com/dviraran/xCell/
xCell生成富集分数,而不是百分比。这不是一种反褶积方法,而是一种富集方法。这意味着它的主要用途是在样本之间进行比较,而不是在细胞类型之间进行比较。
-
产生的score千万不要归一化,否则会造成在这个问题上,不可避免的会导致错误的解释。
-
如果是芯片数据,直接输入表达矩阵即可log之后的
-
如果是RNASeq,可以输入count文件。
ImmuneCellAI
ImmuCellAI(免疫细胞丰度鉴定,abundance identifier)是一个从RNA-Seq和微阵列数据基因表达数据集中估计24种免疫细胞丰度的工具,24种免疫细胞包括18种T细胞和6种其他类型免疫细胞(B细胞,NK细胞,Monocyte细胞,Macrophage细胞,Neutrophil细胞和DC细胞.)
ImmuCellAI还可以评估不同组免疫细胞浸润的差异,预测患者对免疫检查点阻断治疗的反应。
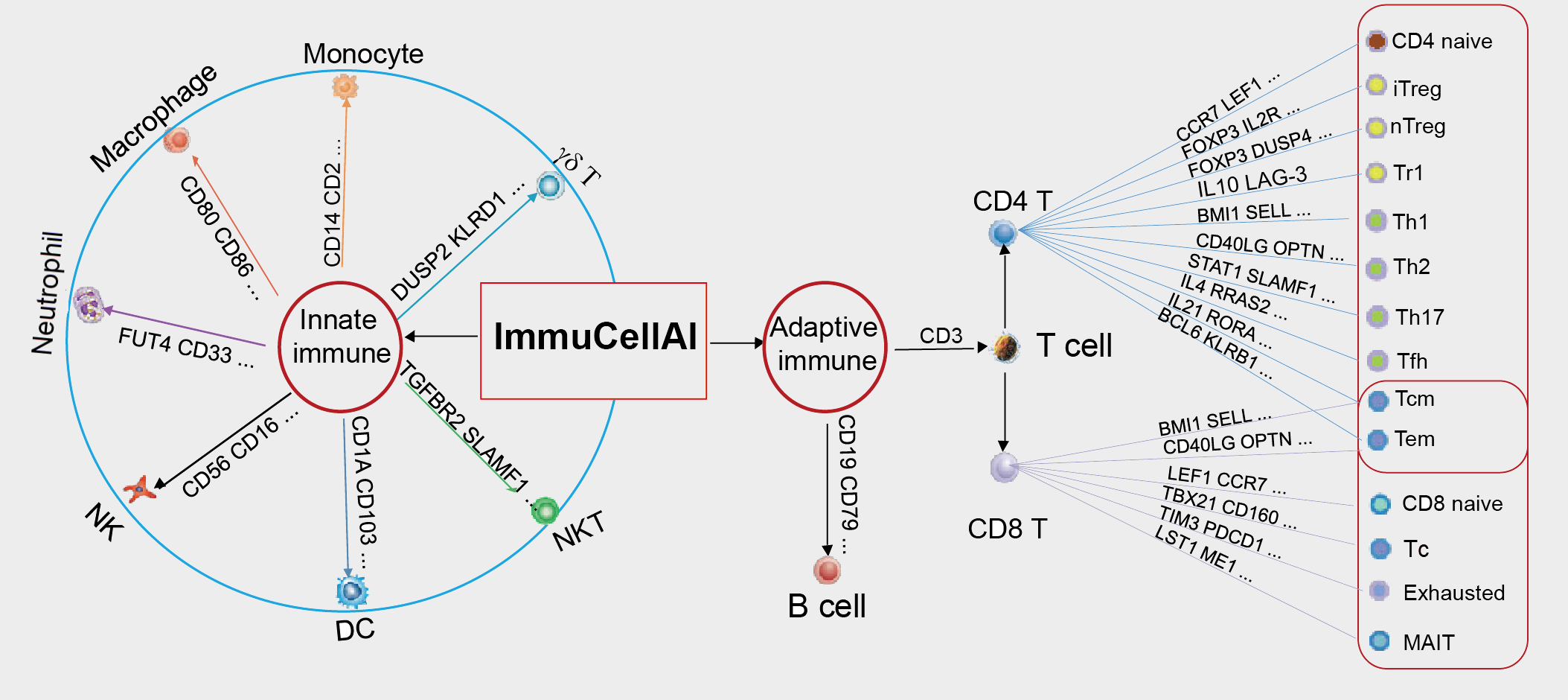
使用方法
-
Analysis模块
- 上传和分析自己的数据
-
Resource模块
- 包括了TCGA数据的免疫浸润分析:癌和癌旁浸润差异的分析,生存分析以及数据的下载。
-
Document模块
- 介绍了网站使用方法。
- 此外,该方法还提供了老鼠的版本:ImmuCellAI-mouse,用以评估小鼠中36种免疫细胞的丰度
-
 image-20210921162355809
image-20210921162355809 - 网站链接:http://bioinfo.life.hust.edu.cn/ImmuCellAI#!/,具体的使用方法,网站也有提供。

quanTIseq
quanTIseq是专门为RNA-seq数据开发的反褶积工具,能够准确定量未知肿瘤的含量,以及量化整体组织的免疫细胞组份。基于约束最小二乘回归和一个新的特征矩阵(来自51个纯化或富集的免疫细胞类型的RNA-seq数据集)。quanTIseq实现了一个完整的反褶积流程来分析RNA-seq数据,能够避免混合物和特征矩阵之间的不一致性。
EPIC
Estimate the Proportion of Immune and Cancer cells (EPIC),用来估计免疫细胞和癌细胞比例。EPIC使用约束最小二乘回归明确地将非负性约束引入反褶积问题,并要求每个样本中所有细胞组分的总和不超过一个。EPIC克服了以往从大量肿瘤基因表达数据预测癌症和免疫细胞或其他非恶性细胞类型的方法的几个局限性,考虑了非特征性和可能高度可变的细胞类型,并在算法上得到了发展。能够广泛应用于大多数实体肿瘤,如黑色素瘤和结直肠样本验证的情况,但不适合血液恶性肿瘤,如白血病或淋巴瘤。
ESTIMATE
- TCGA的各种方法计算的免疫浸润数据可以从TIMER2.0网站上下载(印象笔记里面有附件)
ABIS
DeMixT
转自:Bio Zhong
- 本文固定链接: https://maimengkong.com/kyjc/1468.html
- 转载请注明: : 萌小白 2023年4月22日 于 卖萌控的博客 发表
- 百度已收录
