在 6 月份,基因组学领域的权威期刊Genome Biology发表了一篇方法学评估类文章,在这篇文章中作者系统地分类和评估了目前主要的69种基于全基因组测序(whole genome sequencing,WGS)数据分析结构变异的算法/方法:Comprehensive evaluation of structural variation detection algorithms for whole genome sequencing [1]。
问题
结构变异(structural variation, SV)有哪些类型?
有哪些分析方法/算法可以分析 WGS 数据的结构变异?
如何选择和组合?
导言
如果你已经听说过中国的10万人基因组计划和UK Biobank的50 万人基因组计划就会知道,未来是最不缺全基因组测序数据的。我个人一直相信全基因组测序会在不久的将来成为疾病/药物研究、表型关联分析等领域的首选测序技术。哪怕是截止现在,单单在美国 St. Jude 儿童研究医院的云计算平台就已经托管了超过 11000 例全基因组测序数据样本。我不知道在国内现在是怎样的一番情况。
全基因组测序技术是目前最常见、最具应用前景的高通量测序技术之一。基于这种检测技术,我们可以轻松、便宜地获取包括人类在内的生物或者非生物(如 DNA 和 RNA 病毒)基因组编码信息,并基于这些遗传信息结果来分析和研究物种进化、疾病发生以及药物研发等诸多领域。该技术极大地推动了生物医学、生物信息学/计算生物学、系统生物学等相关学科的发展和融合。
细胞(cell)是生物体的基本组成单元。我们每一个人身上都大约有10^12 - 10^16 数量级的细胞[2]。每一个细胞均包含着我们的全部基因组信息(约 30 亿个碱基对)[3]。无时无刻,我们体内都有大量细胞在消逝,也有大量细胞在生成,即细胞的新陈代谢过程。DNA 复制(replication),即遗传物质(脱氧核酸)从一个细胞被拷贝到了另一个细胞,是其中最常见,最重要,也是必须进行的一个生物学过程。DNA 复制不是 100% 准确无误的,它常常会伴随一些意外的“小变化”和“大变化”,即基因突变。通过实验和计算方法的预测,科研人员发现在癌症中有三分之二的基因突变是来源于 DNA 复制错误 [4,5]。
小变化主要是小片段序列内的改变,主要是两种:单核苷酸变异(single nucleotide varition, SNV)和小片段的序列插入和缺失(insertion and deletion,INDEL)。
大变化即结构变异(structure varition,SV)。SV 常常指的是那些在染色体水平发生了较大片段(大小至少为 50 bp)的改变。染色体数目的改变(倍性变化)一般不包含在结构变异的定义中。
结构变异有哪些类型?
我们假设现在有 A、B、C、D、E 五个基因组区间按顺序排列:ABCDE,以及额外的一个 F 区间,长度分别为 50 bp。以及一段长度大于 50bp 的序列:TTGTG******GGGGG。
- ABCDE → AE 就是发生了 “BCD” 150bp 长度的缺失。
- ABCDE → ABCD**F**E 就是在 D 和 E 之间发生了 50bp 长度的插入。
- 重复区间隔比较小的是串联重复,极端的情况是间隔为0:AAAAAA ,重复区间隔比较大的是散在重复:A*****A*****A。
- 基因序列 TTGTG******GGGGG → GGGGG******GTGTT 就是这个 DNA 序列所在的基因组区间发生了倒位
- 染色体 1 的某个 A 片段接到了染色体 2 的B的后面,就是染色体 1 和染色体 2 发生了染色体异位
综上,SV 主要有以下五种:
1.缺失(deletions,DELs);
2.插入( insertions,INSs)。INSs 主要根据插入序列分为移动元件插入(mobile element insertions,MEIs);线粒体基因组插入(nuclear insertions of mitochondrial genome,NUMTs);病毒序列插入(viral element insertions,VEIs)和其他非特异性插入( insertions of unspecified sequence);
3.重复(duplications,DUPs),DUPs 可以根据重复片段之间的距离分为串联(tandem)和散在(interspersed )重复;
4.倒位(inversions,INVs);
5.异位(translocations,TRAs)
补充:
- 拷贝数变异(copy number variation,CNV)是 DELs 和 INSs 的合称。
- INVs 和 TRAs 不会改变基因组大小,其余均会使得基因组变大或者变小。
分析WGS结构变异的算法
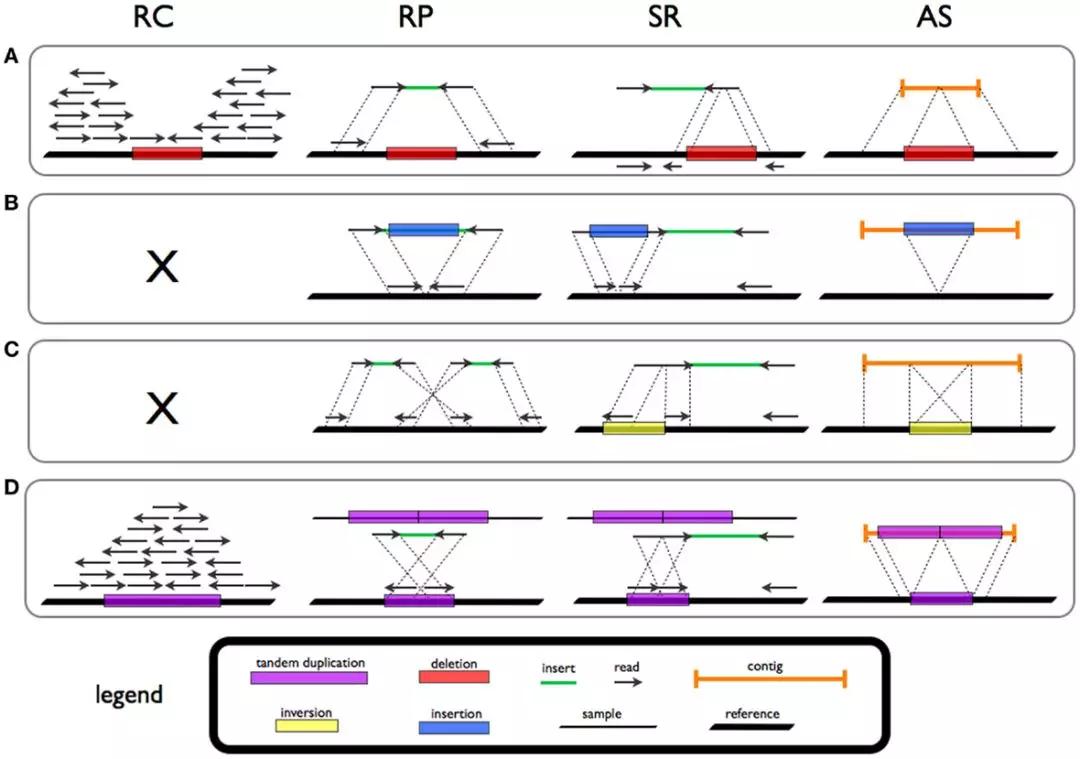
图1 SV 检测策略示意图:A-缺失,B-插入, C-倒位和 D-串联重复;四种分析策略:Read count (RC), Read-pair (RP), Split-read (SR), 和 de novo assembly(AS)[6]
从算法策略来看,目前主要有以下几大类方法可以用于解析 WGS 数据中的结构变异(图1)[7]:
1.Read count (RC) or Read depth (RD),RC 方法假定测序 reads 是随机分布(例如,泊松分布)。我们通过分析 reads 分布的差异,来计算其中的重复和缺失区间。重复的区间将有较高的深度,而缺失区间将有较低的测序深度。
2.Read-pair (RP),RP 方法通过检查配对末端 reads 的长度和方向。配对末端 reads 的距离比预期的要远,表示发生缺失,太近则表示有插入。
3.Split-read (SR),SR 方法可以检测插入(包括移动元素插入)和缺失,分辨率达到单个碱基。它主要是基于 split (soft-clipped) alignment(序列可能来自不同基因组或者同一基因组的不同区间)检测 SV。reads 中的缺口表示发生缺失,参考序列出现不匹配的部分表示插入。
4.De novo assembly (AS),从头 AS 法可以应用于那些精度比较高的测序 reads。不过,由于二代测序 reads 读取的长度限制,会在一定程度上限制该方法的使用。
5.整合上述四种策略,如 RP-SR,RP-RD,RP-AS,RP-SR-AS,RP-SR-RD
上面这些都是分析策略,而基于分析策略/算法之下就是一个个的分析方法:
RD:AS-GENSENG,BICseq2,CNVnator,Control-FREEC, iCopyDAV, OncoSNP-Seq, PennCNV-Seq, readDepth
RP:1-2-3-SV,BreakDancer,Breakway,CLEVER,SVDetect, SVfinder, Ulysses, VariationHunter
SR:Pindel, Socrates, Sprites, SVseq2
AS:FermiKit, laSV, MindTheGap
RP+SR:BreakSeek,DELLY,indelMINER,Meerkat,PRISM, RAPTR, SoftSearch, SoftSV, Wham
RP+RD:forestSV,GASVPro,GenomeSTRiP,inGAP-sv,SoloDel
RP+AS:BASIL-ANISE, Hydra
RP+SR+AS:CREST,GRIDSS,Manta,Pamir,PopIns,SvABA
RP+SR+RD:ERDS,Lumpy,MATCHCLIP,SVelter,TIDDIT
此外还有基于长测序数据(Long Reads,LR)、针对一些特定插入突变(MEI/NUMT/VEI)和其他类型的分析方法
LR: PBHoney-NGM,pbsv,Sniffles
MEI/NUMT/VEI:ITIS,MELT,Mobster,RetroSeq,Tangram,Tea,TEMP,BatVI,HGT-ID,VirusFinder2,VirusSeq
其他:DINUMT,DIGTYPER,SV2,BreakSeq2,MetaSV
用于方法学评估的数据集
我们要测试或者评估一系列生物信息学方法,除了需要筛选哟用于测试的”方法/算法”之外,还需要选择合适的测试数据集(模拟的和真实的)。
作者在这篇文章中使用的测试数据集构成:
1.由 VarSim simulator[8] 联合 ART simulator[9] 产生的短读长模拟数据集:Sim-A 数据集;
2.由 PBSIM[10] 产生的长读长数据集 (–depth = 10, –length-mean = 75,000, –length-sd = 8000):Sim-A-PacBio;
3.其他模拟的数据集(作者开发的脚本构建):Sim-MEI, Sim-NUMT, 和 Sim-VEI;
4.短读长和长读长的真实数据集。
短读长模拟数据集
基于 GRCh37 ,并使用 VarSim simulator 工具构建了一个模拟基因组:引入一系列已知的 SV,包含 50 bp - 1 Mb 大小的 8310 个 SVs (3526 DELs, 1656 DUPs, 2819 INSs, and 309 INVs)。然后,引人 0.1% 基因组大小的 SNP,0.02% 的 Indel 进入父系、母系单倍体基因组。然后再使用 ART simulator 基于该基因组,产生不同读长 (100 bp, 125 bp, 和 150 bp), 插入大小 (400 bp, 500 bp, 和 600 bp), 以及覆盖深度 (10×, 20×, 30×, 和 60×) 的数据集。
Sim-NUMT
UCSC Genome Browser 下载 NUMT 序列(766 个 NumtS 序列)。随机选取其中 200 个 NUMT 序列(至少 100 bp 长)和其他 0.1% 基因组大小的 SNP,0.02% 的 Indel 随机插入到 GRCh37 第十七号染色体。然后再使用 ART simulator 基于该基因组,产生不同读长 (100 bp, 125 bp, 和 150 bp), 插入大小 (400 bp, 500 bp, 和 600 bp), 以及覆盖深度 (10×, 20×, 30×, 和 60×) 的数据集。
Sim-MEI
基于人类一号染色体,使用 BLAST 工具搜索最低 90% 一致、10% 覆盖的 Alu, LINE1, SVA, 和 HERVK 可移动元件 (数量分别为 9548, 1663, 123, 和 10),随机选择其中 651 个序列和其他 0.1% 基因组大小的 SNP,0.02% 的 Indel 随机插入到 GRCh37 第十七号染色体。然后再使用 ART simulator 基于该基因组,产生不同读长 (100 bp, 125 bp, 和 150 bp), 插入大小 (400 bp, 500 bp, 和 600 bp), 以及覆盖深度 (10×, 20×, 30×, 和 60×) 的数据集。
Sim-VEI
NCBI 下载包括疱疹在内的 669 种人类感染性病毒单纯病毒和腺病毒,随机选取其中 100 个序列和其他 0.1% 基因组大小的 SNP,0.02% 的 Indel 随机插入到 GRCh37 第十七号染色体。为了增加序列多样性,再从随机选择的病毒序列区域中提取 500 bp 至 10 kb 长度的片段,并对 0%-5% 的 VEI 序列进行随机替换。然后再使用 ART simulator 基于该基因组,产生不同读长 (100 bp, 125 bp, 和 150 bp), 插入大小 (400 bp, 500 bp, 和 600 bp), 以及覆盖深度 (10×, 20×, 30×, 和 60×) 的数据集。
真实数据集
NA12878 是一个常用作方法学评估的基因组,作者使用的真实数据集主要基于该基因组,并主要来源于:
- Database of Genomic Variants (DGV) 数据库。
- 长读长测序或者基于组装的分析结果
- 其他一些已经发表的、或者实验验证的高可信度的分析结果
处理步骤:
1.作者认为长读长测序或者基于组装的分析结果比 DGV 的结果在短的 DELs 和 INSs 更可信、覆盖的位点也更多,所以移除了 DGV SV 中部分短的 DELs 和 INSs
2.移除 DGV 数据库和长读长测序或者基于组装的分析结果中 >= 95% 重叠的 DELs, DUPs, 和 INVs(> 1kb 的 SV 则设置为具有 >= 90% 以上的重叠)
3.移除 >= 70% 重叠的 DELs
4.移除所有小于 50 bp 的 “SVs” 被移除(除了 INSs)
5.合并 svclassify 研究(PMID:26772178)的高可信的 SVs
6.合并 72 个非冗余、实验验证过的来自于长读长数据的 INV 数据集(PMID:26121404,30001702)
7.和并 InvFEST 数据库(http://invfestdb.uab.cat)
8.提取长度 >30 bp 的 HG00514 SV 结果(ftp://ftp-trace.ncbi.nlm.nih.gov/pub/dbVar/data/Homo_sapiens/by_study/vcf/nstd152.GRCh37.variant_call.vcf.gz), “BND” 被移除,”CNV” 被重新注释为 DEL 和 DUP。
9.提取长度 >30 bp 的 HG002 SV 结果(ftp://ftp-trace.ncbi.nlm.nih.gov//giab/ftp/data/AshkenazimTrio/analysis/NIST_SVs_Integration_v0.6/HG002_SVs_Tier1_v0.6.vcf)
用到的统计学公式
如果我们要深入理解某个生物信息学分析方法的内核,看懂或者理解这些公式的含义是非常重要的。下面我们以其中的三个为例,看一下作者在这篇方法学评估文章中提供的计算公式和统计模型。
首先是经典的准确率和召回率的定义:
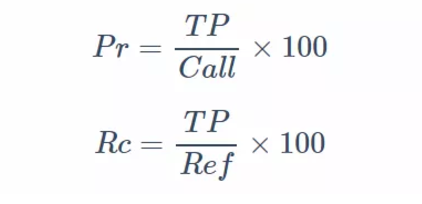
注释:Pr (准确度),Precision 的简写;Rc (召回率),Recall 的简写;TP(真阳性位点数),Call(检测出的 SV 位点数),Ref(所有已知的 SV 位点数)
准确率(Pr)和召回率(Rc)是评估生物信息学分析方法的必备参数,也是生物信息学方法学论文中常出现的描述:如本方法使用样本量为 n 的某类数据建立了一个某某模型/算法,并利用某某数据集进行了评估,发现本方法的准确率为 90%,召回率为 70%,与同类方法相比分别提升了百分之 50%。
准确率在这篇文章里面我们可以理解为每种 SV 检测方法得到的确定为“真“的 SV 位点数目分别除以每种方法所有检测出的位点数,再分别乘上 100 化为百分数。举个例子:A 方法检出 100 个位点,其中 50 个为”真“,那么 A 方法的准确率为 50 %。
召回率即每种 SV 检测方法得到的确定为“真“的 SV 位点数目分别除以全部已知为”真“位点。举个例子(注意和上个例子进行对比):A方法检出 100 个位点,而已知 A 方法测试的数据集总共有 200 个 SV 位点,那么 A 方法的召回率为 50%。

作者为了确定多方法联用分析策略对不同 SV 类型在模拟数据(sim)和真实数据(real)的整合准确度,提出了一个整合准确率分数(combined Pr,cPr),mPr 是 mean Precision Score 的缩写,即平均准确度。我们如何理解这个公式呢?首先是看变化趋势,模拟数据和真实数据的 Pr 值越大,那么 cPr 就越大,反之则越小。另外,这里分子和分母都用的是乘法,所以这两个因子是互相影响的,即 Pr (sim) 和Pr (real) 的任意一个数值很小时,cPr 仍然会很小。如果我们换成加号的话,当公式中分子的某个值很大,另一个值很小时, cPr 并不会变得很小,而会约等于较大值。
我们假设 A,B,C 在模拟数据的准确率分别为 10%,20%,70%,在真实数据的准确率分别为 5%,15%,50%。mPr (sim) 为 (10 + 20 + 70) / 3 = 33.33%,mPr (real)为(5 + 15 + 50) / 3 = 23.33%。A 的 cPr(就像你的考试分数) 为 100 * ( 10 * 5 ) / (33.33 * 23.33) = 6.43,B 的 cPr 为 100 * (20 * 15) / (33.33 * 23.33) = 38.58,C 的 cPr 为 100 * (70 * 50) / (33.33 * 23.33) = 450.10。所以使用上面这个公式可以计算得到 A,B,C 三种方法的 cPr 分数为别为 6.43,38.58,450.10。从上面这个 A、B、C 的例子我们可以知道这个公式的分母是一个由 A、B、C 在模拟数据和真实数据计算出得到的准确度共同决定的一个常量,我们可以理解为等比例的缩小了这个评估分数。
另外,作者又提出了一种 “Modified F-measures” 统计方法(多种统计参数的结合)来评估单个数据集中各种类型 SV 算法的敏感度和准确度,这个值越大,表明这个方法越”好“。公式如下:
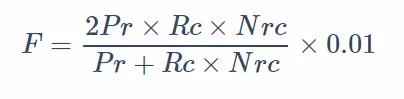
这里的 Nrc(normalized index of Recall) 是一个根据 SV 类型变化的常量:DEL, DUP, INS, 和 INV 分别为 2.9, 4.0, 2.4, 和 2.4。它的主要作用是矫正模拟数据和真实数据在召回率上由模拟数据参考很多真实数据 SVs 结果引起的差异(真实数据的 recall 系统性地小于模拟数据)。
如何选择和组合
生物信息学方法评估类文章为我们选择和组合数据分析方法提供了非常重要的参考依据。这篇文章的作者基于模拟数据和真实数据集对不同类型的 SV 以及不同的数据分析方法作的一系列评估的结果,囊括了我们关心的几个方法学选择的重要因素,如准确率、召回率、变异大小的影响、运行时间和内存占用等。
GRIDSS, Lumpy, SVseq2, SoftSV, Manta, 和 Wham 是作者在摘要中明确点名出来表现比较好的算法。作者也公布了用于测试的代码,你可以在自己的数据集进行测试后根据结果进行最终的方案选择:stat-lab/EvalSVcallers
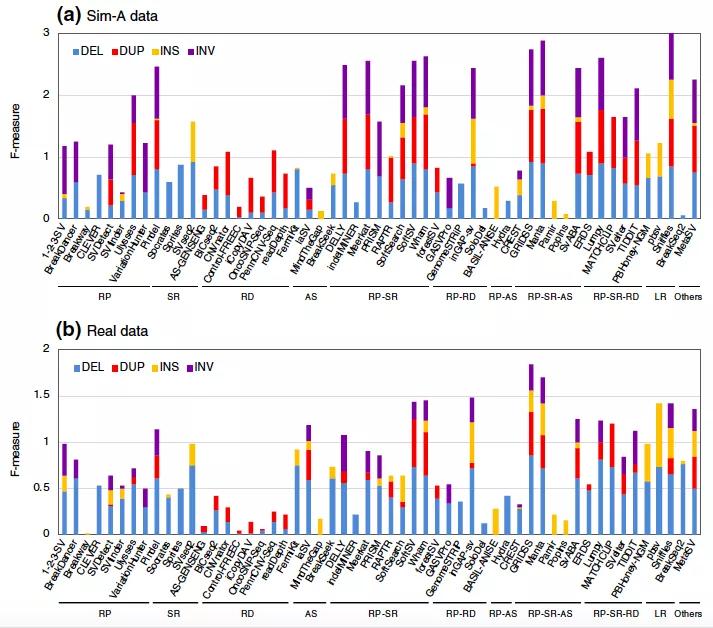
图2 不同 SV 分析方法在 DELs, DUPs, INSs, 和 INVs 的表现(主要是评估敏感度和准确度),simulated (a) and the NA12878 real data (b).
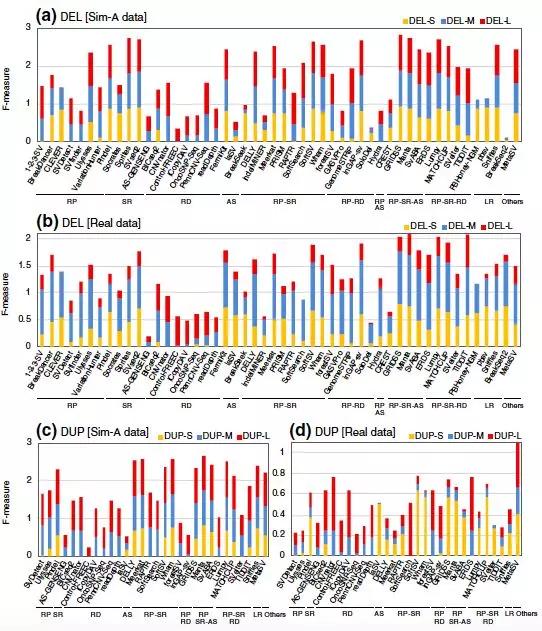
图3 不同 SV 分析方法在不同大小(短、中、长)下的 DELs 和 DUPs 的表现
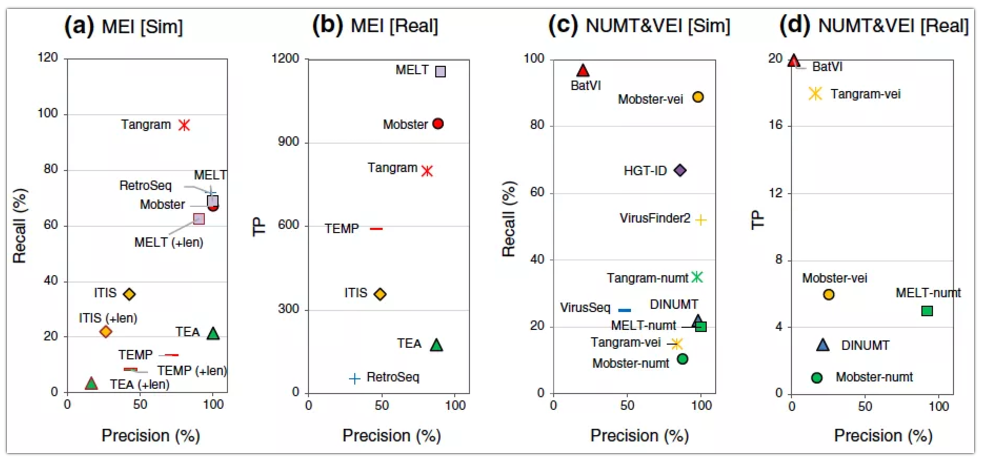
图4 MEI/NUMT/VEI (插入)分析算法评估
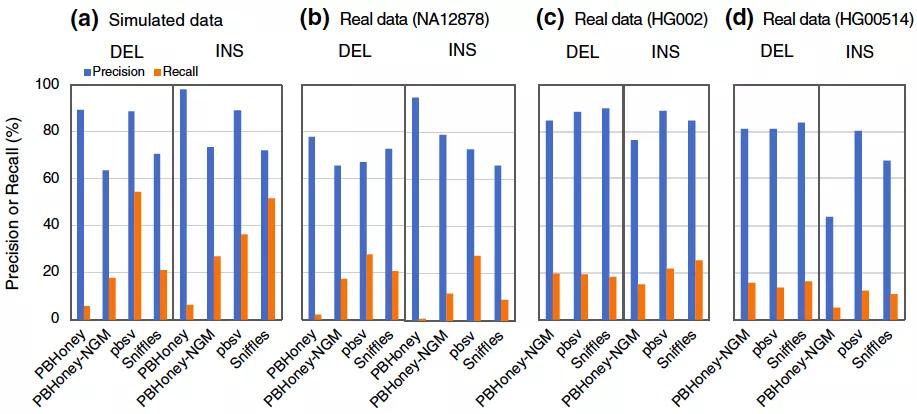
图5 基于Long reads (LR)方法的准确度和召回率评估
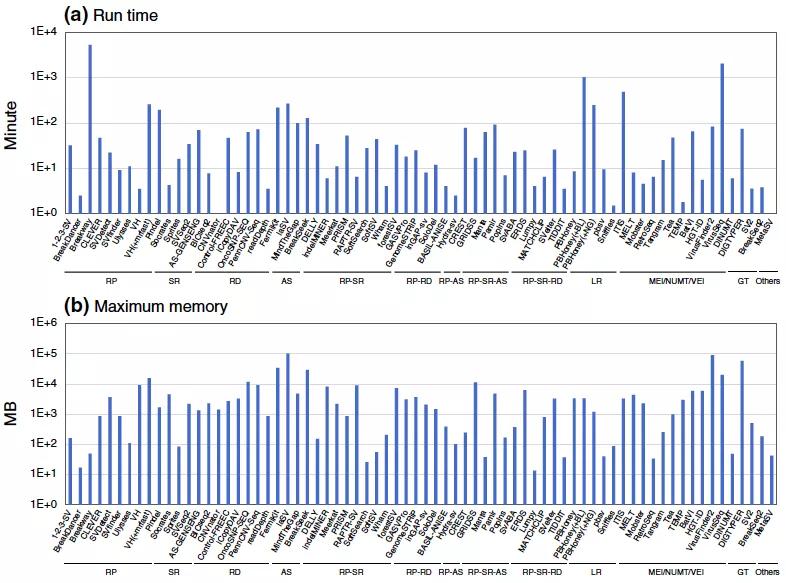
图6 SV 分析方法计算时间和内存消耗评估
参考资料
[1] Comprehensive evaluation of structural variation detection algorithms for whole genome sequencing. Genome Biol. 2019 Jun 3;20(1):117. doi: 10.1186/s13059-019-1720-5.
[2] An estimation of the number of cells in the human body. Ann Hum Biol. 2013 Nov-Dec;40(6):463-71. doi: 10.3109⁄03014460.2013.807878. Epub 2013 Jul 5
[3] .碱基又称核碱基、含氮碱基,是一种含氮化合物,且可以通过氢键形成碱基对。它是生物遗传物质(核糖核酸,RNA/脱氧核糖核酸,DNA)的基本组成元件之一。
[4] Tomasetti, C. & Vogelstein, B. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 347, 78–81 (2015).
[5] Tomasetti, C., Li, L. & Vogelstein, B. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science 355, 1330–1334 (2017).
[6] Detection of genomic structural variants from next-generation sequencing data. Frontiers in Bioengineering and Biotechnology. 3 (92): 92. doi:10.3389/fbioe.2015.00092. PMC 4479793. PMID 26161383.
[7] https://en.wikipedia.org/wiki/Structural_variation
[8] http://bioinform.github.io/varsim/
[9] https://www.niehs.nih.gov/research/resources/software/art
[10] https://code.google.com/archive/p/pbsim
来源于网络,侵权请联系删除。
- 本文固定链接: https://maimengkong.com/zu/1390.html
- 转载请注明: : 萌小白 2023年3月3日 于 卖萌控的博客 发表
- 百度已收录
