2022
05-03
05-03
生信必会的SAM格式,该怎么看?
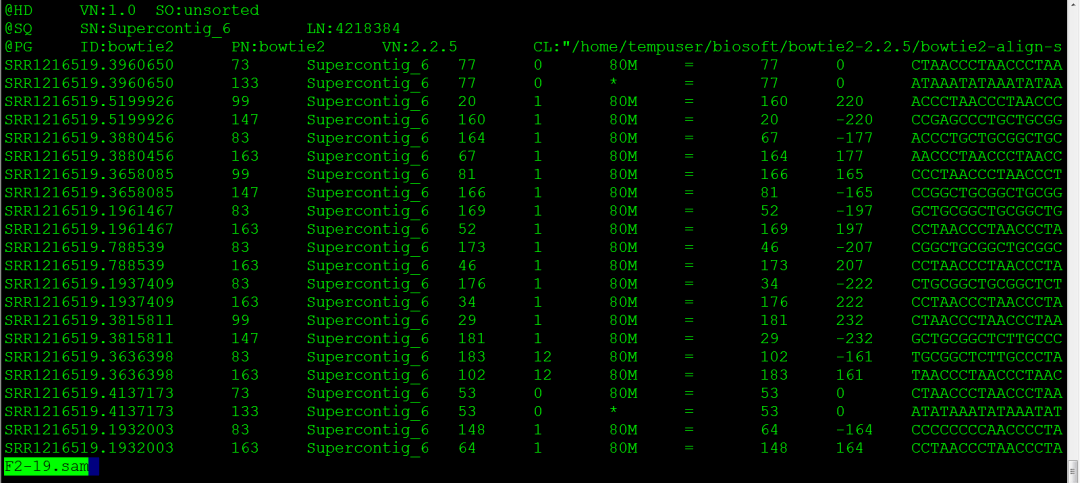 对高通量测序数据进行比对,就是将测序得到的reads定位到基因组序列上,对illumina或454得到的short reads比对的软件主要有Bowtie BWA HISAT Tophat。SAM格式,是序列比对文件的格式。分为头部区和主体区,都以tab分列。@HD VN:1.0 SO:unsorted头部区第一行,VN是格式版本,SO是比对的类型,有unknown,unsorted,q...阅读... 阅 读 全 部 >
对高通量测序数据进行比对,就是将测序得到的reads定位到基因组序列上,对illumina或454得到的short reads比对的软件主要有Bowtie BWA HISAT Tophat。SAM格式,是序列比对文件的格式。分为头部区和主体区,都以tab分列。@HD VN:1.0 SO:unsorted头部区第一行,VN是格式版本,SO是比对的类型,有unknown,unsorted,q...阅读... 阅 读 全 部 >
 如果你对生物信息不那么了解,或许会搞混FASTQ和FASTA,它们都是序列保存的一种格式,是用于NGS分析的基础数据。其实二者很好区分,并且可以通过一定的处理进行格式转换。FASTQFASTQ:是基于文本的,保存生物序列(通常是核酸序列)和其测序质量信息的标准格式。你拿到的测序数据均是fastq格式的。其序列以及质量信息都是使用一个ASCII字符标示,最初是由Sanger开发,目的是将F...阅读...
如果你对生物信息不那么了解,或许会搞混FASTQ和FASTA,它们都是序列保存的一种格式,是用于NGS分析的基础数据。其实二者很好区分,并且可以通过一定的处理进行格式转换。FASTQFASTQ:是基于文本的,保存生物序列(通常是核酸序列)和其测序质量信息的标准格式。你拿到的测序数据均是fastq格式的。其序列以及质量信息都是使用一个ASCII字符标示,最初是由Sanger开发,目的是将F...阅读...  生信分析过程中,会与很多不同格式的文件打交道,除了原始测序数据 fastq 之外,还需要准备基因组文件 fasta 格式和基因注释文件 gtf 格式。在分析的过程中还会有众多中间文件的生成,如 bed 、 bed12 、 sam 、 bam 、 wig 、 bigwig 、 bedgraph 等,生成后我们一般会查看下内容了解文件每一列的含义,以此来决定需要提取哪些有用信息列来进行下...阅读全文...
生信分析过程中,会与很多不同格式的文件打交道,除了原始测序数据 fastq 之外,还需要准备基因组文件 fasta 格式和基因注释文件 gtf 格式。在分析的过程中还会有众多中间文件的生成,如 bed 、 bed12 、 sam 、 bam 、 wig 、 bigwig 、 bedgraph 等,生成后我们一般会查看下内容了解文件每一列的含义,以此来决定需要提取哪些有用信息列来进行下...阅读全文... 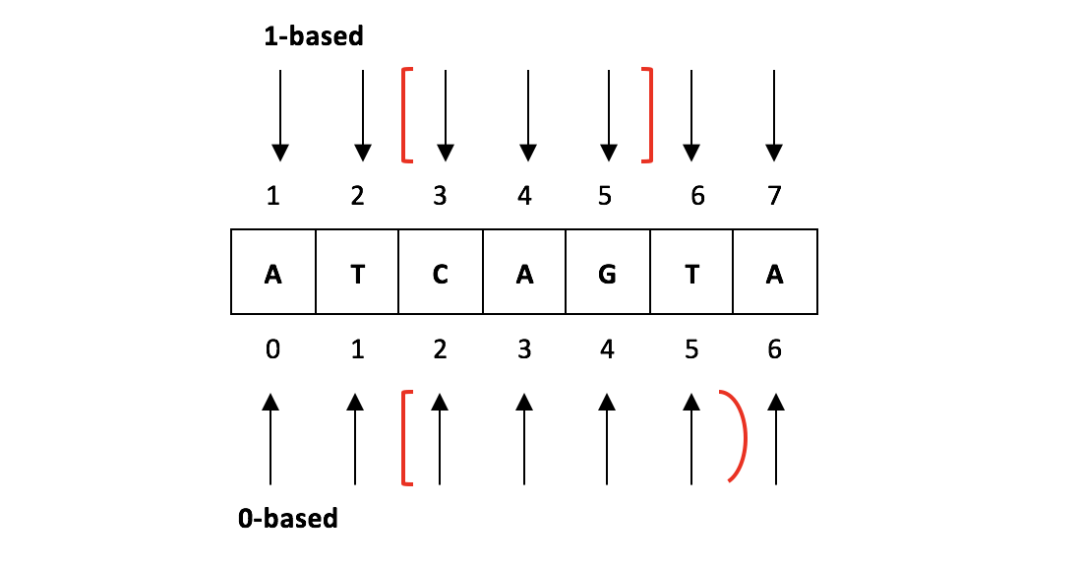 生信分析过程中,会与很多不同格式的文件打交道,除了原始测序数据 fastq 之外,还需要准备基因组文件 fasta 格式和基因注释文件 gtf 格式。在分析的过程中还会有众多中间文件的生成,如 bed 、 bed12 、 sam 、 bam 、 wig 、 bigwig 、 bedgraph 等,生成后我们一般会查看下内容了解文件每一列的含义,以此来决定需要提取哪些有用信息列来进行下...阅读全文...
生信分析过程中,会与很多不同格式的文件打交道,除了原始测序数据 fastq 之外,还需要准备基因组文件 fasta 格式和基因注释文件 gtf 格式。在分析的过程中还会有众多中间文件的生成,如 bed 、 bed12 、 sam 、 bam 、 wig 、 bigwig 、 bedgraph 等,生成后我们一般会查看下内容了解文件每一列的含义,以此来决定需要提取哪些有用信息列来进行下...阅读全文... 