2024
05-01
05-01
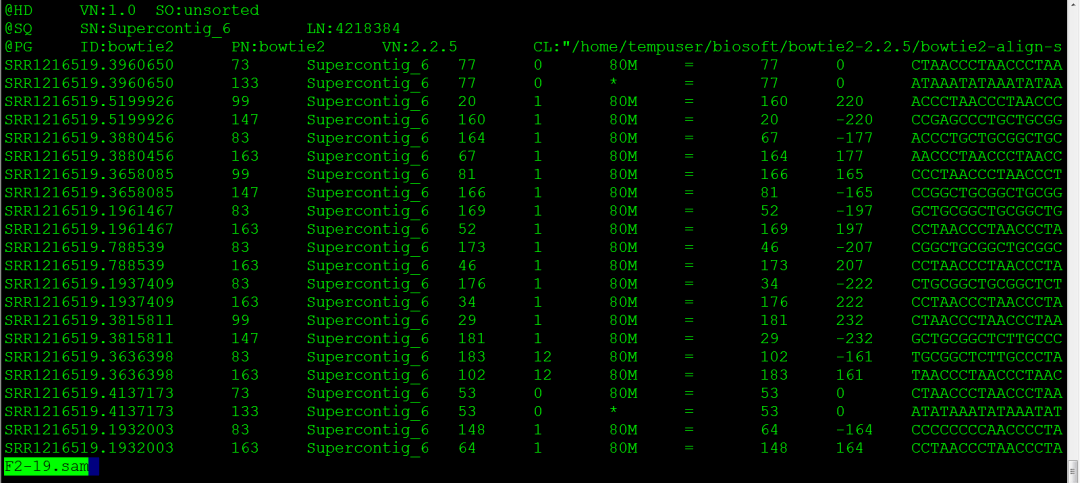 对高通量测序数据进行比对,就是将测序得到的reads定位到基因组序列上,对illumina或454得到的short reads比对的软件主要有Bowtie BWA HISAT Tophat。SAM格式,是序列比对文件的格式。分为头部区和主体区,都以tab分列。@HD VN:1.0 SO:unsorted头部区第一行,VN是格式版本,SO是比对的类型,有unknown,unsorted,q...阅读... 阅 读 全 部 >
对高通量测序数据进行比对,就是将测序得到的reads定位到基因组序列上,对illumina或454得到的short reads比对的软件主要有Bowtie BWA HISAT Tophat。SAM格式,是序列比对文件的格式。分为头部区和主体区,都以tab分列。@HD VN:1.0 SO:unsorted头部区第一行,VN是格式版本,SO是比对的类型,有unknown,unsorted,q...阅读... 阅 读 全 部 >
 如果你对生物信息不那么了解,或许会搞混FASTQ和FASTA,它们都是序列保存的一种格式,是用于NGS分析的基础数据。其实二者很好区分,并且可以通过一定的处理进行格式转换。FASTQFASTQ:是基于文本的,保存生物序列(通常是核酸序列)和其测序质量信息的标准格式。你拿到的测序数据均是fastq格式的。其序列以及质量信息都是使用一个ASCII字符标示,最初是由Sanger开发,目的是将F...阅读... 阅 读 全 部 >
如果你对生物信息不那么了解,或许会搞混FASTQ和FASTA,它们都是序列保存的一种格式,是用于NGS分析的基础数据。其实二者很好区分,并且可以通过一定的处理进行格式转换。FASTQFASTQ:是基于文本的,保存生物序列(通常是核酸序列)和其测序质量信息的标准格式。你拿到的测序数据均是fastq格式的。其序列以及质量信息都是使用一个ASCII字符标示,最初是由Sanger开发,目的是将F...阅读... 阅 读 全 部 >
 生信分析过程中,会与很多不同格式的文件打交道,除了原始测序数据 fastq 之外,还需要准备基因组文件 fasta 格式和基因注释文件 gtf 格式。在分析的过程中还会有众多中间文件的生成,如 bed 、 bed12 、 sam 、 bam 、 wig 、 bigwig 、 bedgraph 等,生成后我们一般会查看下内容了解文件每一列的含义,以此来决定需要提取哪些有用信息列来进行下...阅读全文... 阅 读 全 部 >
生信分析过程中,会与很多不同格式的文件打交道,除了原始测序数据 fastq 之外,还需要准备基因组文件 fasta 格式和基因注释文件 gtf 格式。在分析的过程中还会有众多中间文件的生成,如 bed 、 bed12 、 sam 、 bam 、 wig 、 bigwig 、 bedgraph 等,生成后我们一般会查看下内容了解文件每一列的含义,以此来决定需要提取哪些有用信息列来进行下...阅读全文... 阅 读 全 部 >
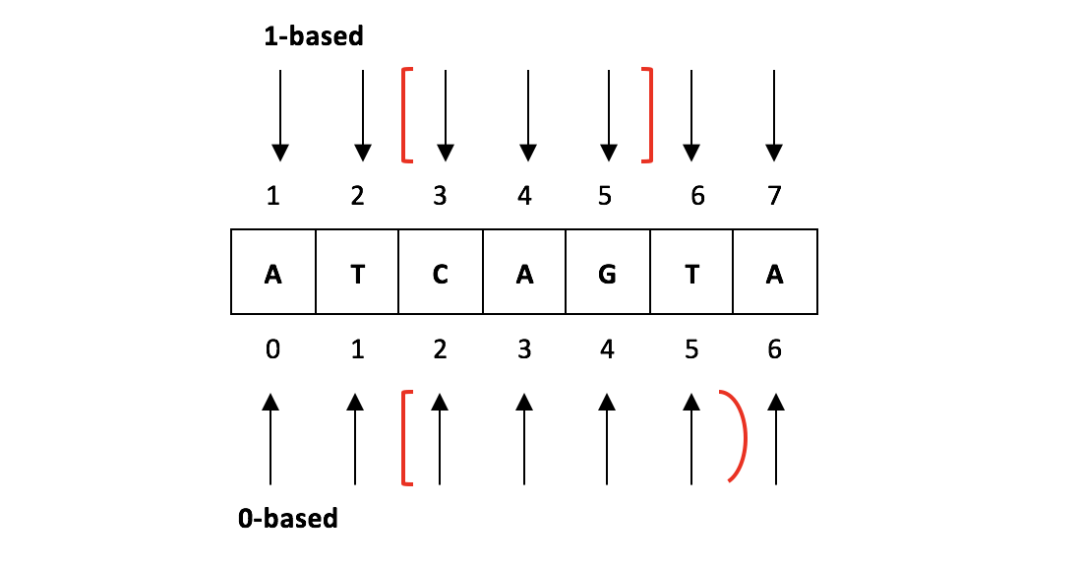 生信分析过程中,会与很多不同格式的文件打交道,除了原始测序数据 fastq 之外,还需要准备基因组文件 fasta 格式和基因注释文件 gtf 格式。在分析的过程中还会有众多中间文件的生成,如 bed 、 bed12 、 sam 、 bam 、 wig 、 bigwig 、 bedgraph 等,生成后我们一般会查看下内容了解文件每一列的含义,以此来决定需要提取哪些有用信息列来进行下...阅读全文... 阅 读 全 部 >
生信分析过程中,会与很多不同格式的文件打交道,除了原始测序数据 fastq 之外,还需要准备基因组文件 fasta 格式和基因注释文件 gtf 格式。在分析的过程中还会有众多中间文件的生成,如 bed 、 bed12 、 sam 、 bam 、 wig 、 bigwig 、 bedgraph 等,生成后我们一般会查看下内容了解文件每一列的含义,以此来决定需要提取哪些有用信息列来进行下...阅读全文... 阅 读 全 部 >
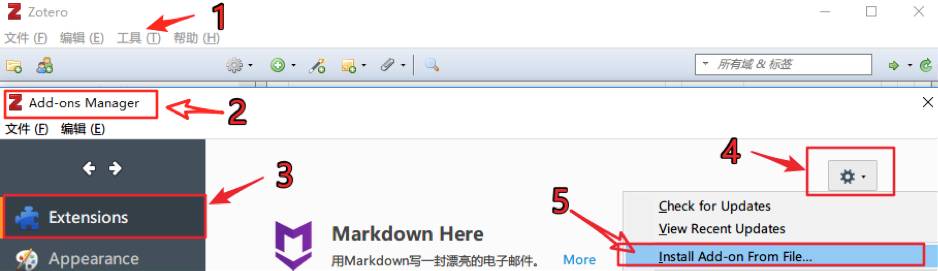 一、前言 之前有说过,Zotero一大好处就是开源,免费。而软件开源的好处是,会有很多技术大拿一起再GitHub上面给软件写各种各样强大的插件。Zotero就是如此,借助于插件, Zotero战斗能力倍增,可以让大家更为轻松自如地学习与管理文献。今天,小编就为大家带来Zotfile这个插件,感受下开源软件的人性化设计。 二、Zotfile 插件的使用 2.1 安装 直接进入Zotfile... 阅 读 全 部 >
一、前言 之前有说过,Zotero一大好处就是开源,免费。而软件开源的好处是,会有很多技术大拿一起再GitHub上面给软件写各种各样强大的插件。Zotero就是如此,借助于插件, Zotero战斗能力倍增,可以让大家更为轻松自如地学习与管理文献。今天,小编就为大家带来Zotfile这个插件,感受下开源软件的人性化设计。 二、Zotfile 插件的使用 2.1 安装 直接进入Zotfile... 阅 读 全 部 >
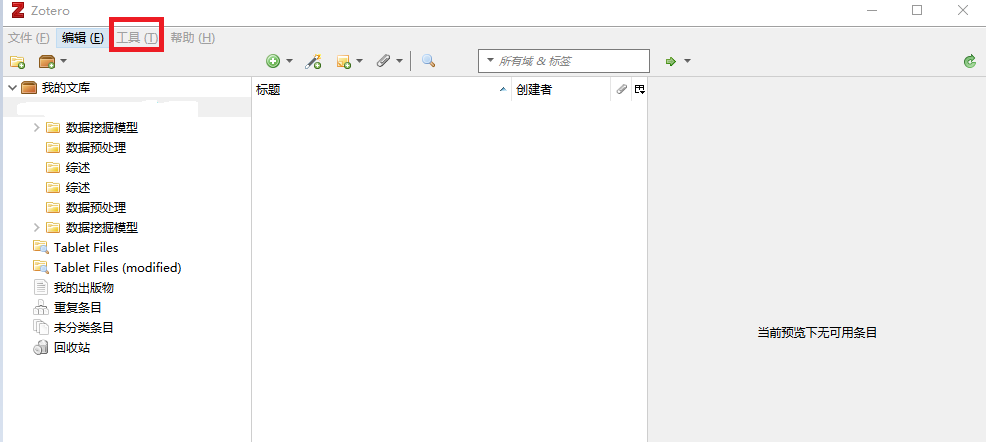 1.安装ZotFile插件*** 以后补充 ***2.配置ZotFile 配置 3.整理操作(1)将文件拖进Zotero软件相应的目录(自己创建) (2)查看文件位置 (未整理之前) (3)整理 (实质是拷贝了一个新文件,按照命名规则对文件进行了重命名,并存储到了自定义的目录下如:本配置中的 “E:\workspace\Zotero\Paper... 阅读... 阅 读 全 部 >
1.安装ZotFile插件*** 以后补充 ***2.配置ZotFile 配置 3.整理操作(1)将文件拖进Zotero软件相应的目录(自己创建) (2)查看文件位置 (未整理之前) (3)整理 (实质是拷贝了一个新文件,按照命名规则对文件进行了重命名,并存储到了自定义的目录下如:本配置中的 “E:\workspace\Zotero\Paper... 阅读... 阅 读 全 部 >
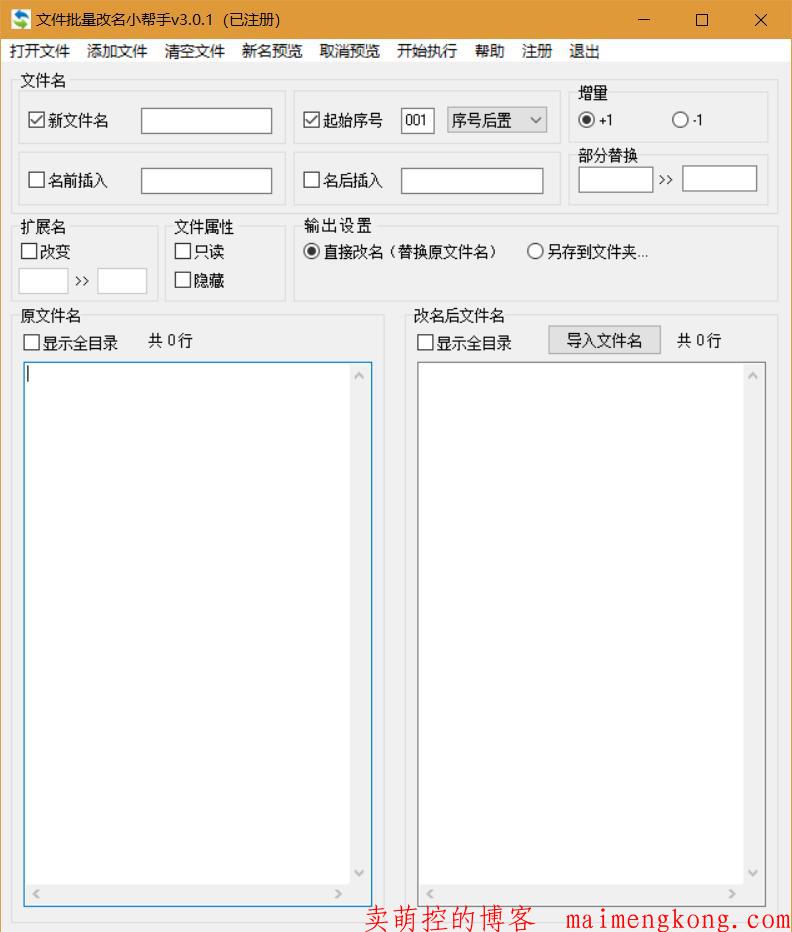 快捷、方便、直观的实现文件批量改名。 支持所有文件格式;支持自动排列序号;支持改变扩展名(后缀名);支持修改文件属性(只读、隐藏文件);支持文件名部分替换;支持在原文件名前插入字符;支持在原文件名后插入字符;支持改名后另存到指定文件夹,保留原文件;支持预览; 文件批量改名v3....阅读全文>>... 阅 读 全 部 >
快捷、方便、直观的实现文件批量改名。 支持所有文件格式;支持自动排列序号;支持改变扩展名(后缀名);支持修改文件属性(只读、隐藏文件);支持文件名部分替换;支持在原文件名前插入字符;支持在原文件名后插入字符;支持改名后另存到指定文件夹,保留原文件;支持预览; 文件批量改名v3....阅读全文>>... 阅 读 全 部 >
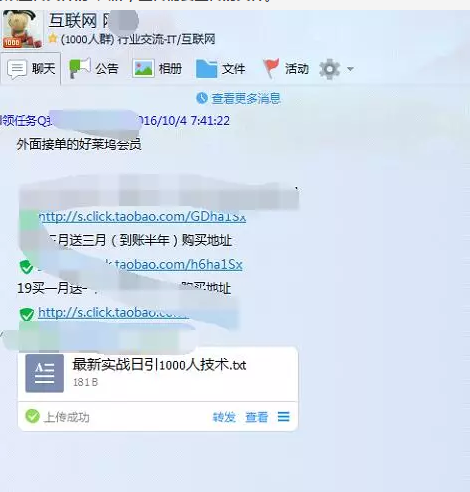 这两天“圈内”都流传着QQ群文件破群规无限制上传的漏洞。可以实现无限制的在任何群内上传任何文件,包括设置了禁止普通群成员上传文件的QQ群。且可以实现上传文件后“隐身”,不被群主或管理员发现。标题或许有些夸张,但博主觉得还比较实用,故分享给大家,希望对大家有所帮助。具体操作步骤如下:1、找到可以上传文件的QQ群,上传需要上传的文件。2、在已上传QQ群点击“转发”,转发可以同时转发...阅读全文>... 阅 读 全 部 >
这两天“圈内”都流传着QQ群文件破群规无限制上传的漏洞。可以实现无限制的在任何群内上传任何文件,包括设置了禁止普通群成员上传文件的QQ群。且可以实现上传文件后“隐身”,不被群主或管理员发现。标题或许有些夸张,但博主觉得还比较实用,故分享给大家,希望对大家有所帮助。具体操作步骤如下:1、找到可以上传文件的QQ群,上传需要上传的文件。2、在已上传QQ群点击“转发”,转发可以同时转发...阅读全文>... 阅 读 全 部 >

生信圈
欢迎您支持我的公众号
点击此处可关闭
 阅读全文>>...
阅读全文>>...  阅读全文>>...
阅读全文>>...