2022
01-22
01-22
进化树的构建与美化
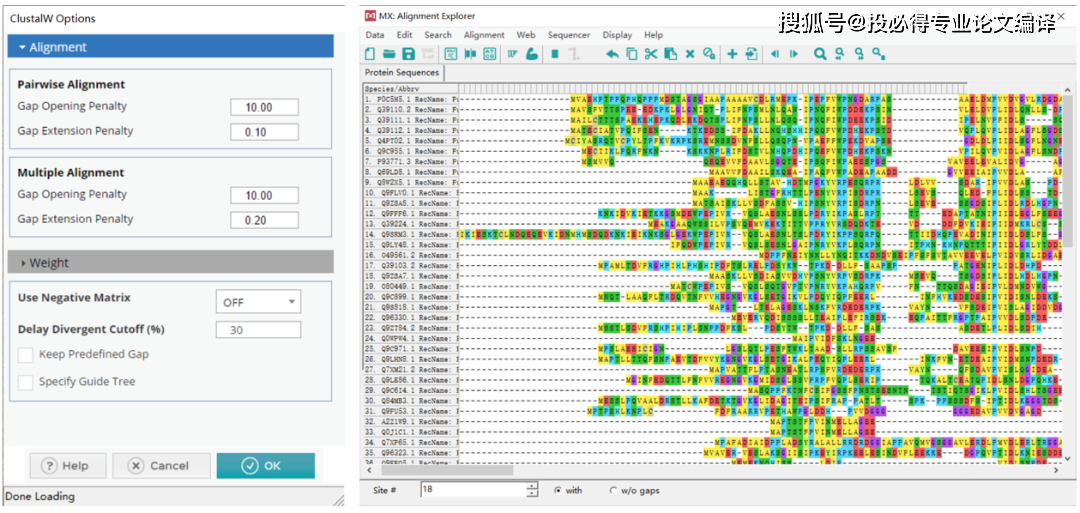 1.进化树1.0版本上一期已经给大家介绍根据基因或者蛋白ID在NCBI上进行BLAST并下载FASTA序列,然后将序列导入MEGA软件中进行比对,最后生成.meg的序列比对文件。那么用这个文件是不是就能构建进化树呢,当然可以。方法也很简单,单击菜单【File】→【Open A File/Session】,打开上述.meg文件,点击工具栏中的【PHYLOGENY】→【C...阅读全文>>... 阅 读 全 部 >
1.进化树1.0版本上一期已经给大家介绍根据基因或者蛋白ID在NCBI上进行BLAST并下载FASTA序列,然后将序列导入MEGA软件中进行比对,最后生成.meg的序列比对文件。那么用这个文件是不是就能构建进化树呢,当然可以。方法也很简单,单击菜单【File】→【Open A File/Session】,打开上述.meg文件,点击工具栏中的【PHYLOGENY】→【C...阅读全文>>... 阅 读 全 部 >
1.进化树1.0版本上一期已经给大家介绍根据基因或者蛋白ID在NCBI上进行BLAST并下载FASTA序列,然后将序列导入MEGA软件中进行比对,最后生成.meg的序列比对文件。那么用这个文件是不是就能构建进化树呢,当然可以。方法也很简单,单击菜单【File】→【Open A File/Session】,打开上述.meg文件,点击工具栏中的【PHYLOGENY】→【C...阅读全文>>... 阅 读 全 部 >
1.进化树1.0版本上一期已经给大家介绍根据基因或者蛋白ID在NCBI上进行BLAST并下载FASTA序列,然后将序列导入MEGA软件中进行比对,最后生成.meg的序列比对文件。那么用这个文件是不是就能构建进化树呢,当然可以。方法也很简单,单击菜单【File】→【Open A File/Session】,打开上述.meg文件,点击工具栏中的【PHYLOGENY】→【C...阅读全文>>... 阅 读 全 部 >
 一、查找基因序列、mRNA序列进入NCBI 主页,在 search 后面选择 Gene,输入需要查找的基因的名字,点击search,查看结果。以基因P53为例,搜索结果如图:点击红框部分,进入并下拉,可以看到大量的信息,如下图:二、用Probe查找已经公布的引物序列进入NCBI主页,在下拉菜单选择Probe之后填写需要查找的基因名称。...阅读全文>>... 阅 读 全 部 >
一、查找基因序列、mRNA序列进入NCBI 主页,在 search 后面选择 Gene,输入需要查找的基因的名字,点击search,查看结果。以基因P53为例,搜索结果如图:点击红框部分,进入并下拉,可以看到大量的信息,如下图:二、用Probe查找已经公布的引物序列进入NCBI主页,在下拉菜单选择Probe之后填写需要查找的基因名称。...阅读全文>>... 阅 读 全 部 >
 导读:随着二代测序技术临床应用的不断增加,越来越多与癌症发生发展密切相关的突变被鉴定出来。将基因突变的结果更好地转化为实际临床应用,统一而通用的突变命名规则就显得尤为重要。人类基因组变异协会(HGVS:Human Genome Variation Society)规则是目前学术界所公认的命名规则。从不同的维度出发,相同的基因突变可以有多种不同的表现形式,例如,参考序列的不同、表...阅读全文>... 阅 读 全 部 >
导读:随着二代测序技术临床应用的不断增加,越来越多与癌症发生发展密切相关的突变被鉴定出来。将基因突变的结果更好地转化为实际临床应用,统一而通用的突变命名规则就显得尤为重要。人类基因组变异协会(HGVS:Human Genome Variation Society)规则是目前学术界所公认的命名规则。从不同的维度出发,相同的基因突变可以有多种不同的表现形式,例如,参考序列的不同、表...阅读全文>... 阅 读 全 部 >
 如果你对生物信息不那么了解,或许会搞混FASTQ和FASTA,它们都是序列保存的一种格式,是用于NGS分析的基础数据。其实二者很好区分,并且可以通过一定的处理进行格式转换。FASTQFASTQ:是基于文本的,保存生物序列(通常是核酸序列)和其测序质量信息的标准格式。你拿到的测序数据均是fastq格式的。其序列以及质量信息都是使用一个ASCII字符标示,最初是由Sanger开发,目的是将F...阅读... 阅 读 全 部 >
如果你对生物信息不那么了解,或许会搞混FASTQ和FASTA,它们都是序列保存的一种格式,是用于NGS分析的基础数据。其实二者很好区分,并且可以通过一定的处理进行格式转换。FASTQFASTQ:是基于文本的,保存生物序列(通常是核酸序列)和其测序质量信息的标准格式。你拿到的测序数据均是fastq格式的。其序列以及质量信息都是使用一个ASCII字符标示,最初是由Sanger开发,目的是将F...阅读... 阅 读 全 部 >
 上周的干货软文为您介绍了如何在Ensembl数据库查找目标基因序列(Ensembl篇),这周我们将进入NCBI篇,为您讲解如何在该数据库查找目标基因序列。搜索已被RefSeq收录的基因序列NCBI,是美国国立生物技术信息中心(National Center for Biotechnology Information)的英文缩写。与专攻基因组检索的Ensembl不同,NCBI数据...阅读全文>... 阅 读 全 部 >
上周的干货软文为您介绍了如何在Ensembl数据库查找目标基因序列(Ensembl篇),这周我们将进入NCBI篇,为您讲解如何在该数据库查找目标基因序列。搜索已被RefSeq收录的基因序列NCBI,是美国国立生物技术信息中心(National Center for Biotechnology Information)的英文缩写。与专攻基因组检索的Ensembl不同,NCBI数据...阅读全文>... 阅 读 全 部 >

生信圈
欢迎您支持我的公众号
点击此处可关闭