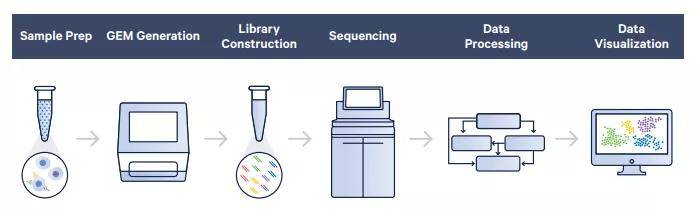
单细胞RNA测序(scRNA-seq)是一个新兴的测序技术,通过解析组织中单个细胞的基因表达水平,来研究细胞胞间异质性、区分细胞类型和亚型、识别细胞类型特异性基因及揭示细胞动态过程等。
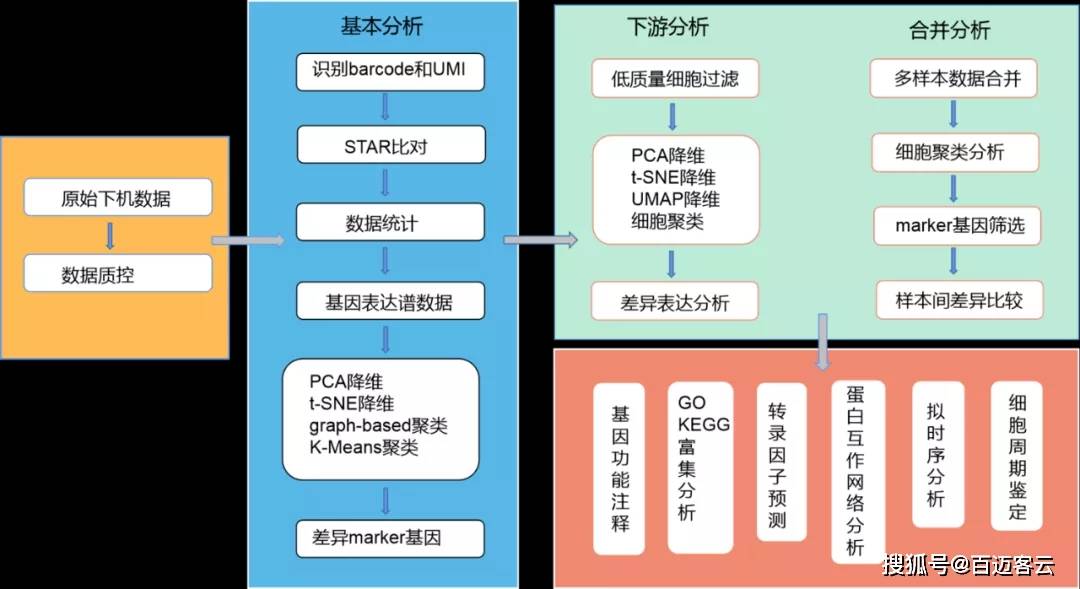
百迈客利用10X Genomics官方软件CellRanger进行数据比对、基因定量以及细胞鉴定。同时cellranger还基于鉴定的细胞进行了细胞亚群分析以及差异表达基因分析。根据cellranger鉴定出的细胞类型以及定量结果,后续采用Seurat软件对数据做了二次过滤,对过滤后的数据进行二次分析,包括:细胞亚群分析、差异表达基因分析、GO/KEGG富集分析以及PPI网络分析等等。
一、数据比对及基因定量
CellRanger是10X Genomics官方推荐的用于单细胞转录组分析的软件。目前使用的版本为:cellranger-6.0.0。Cellranger软件的下载安装可以参考10X官方网站:https://www.10xgenomics.com/products。参考基因组的选择需根据物种选择,人类样品推荐选择GRCh38_release95,小鼠样品推荐选择GRCm38_release95。除了上述推荐的参考基因组,百迈客还为老师准备了其他版本的参考基因组选择。
Barcode用来区分细胞,UMI用来区分转录本。主要用于获得细胞基因表达的矩阵。Fraction Reads in Cells≥70%,Reads Mapped Confidently to Transcriptome≥60%。
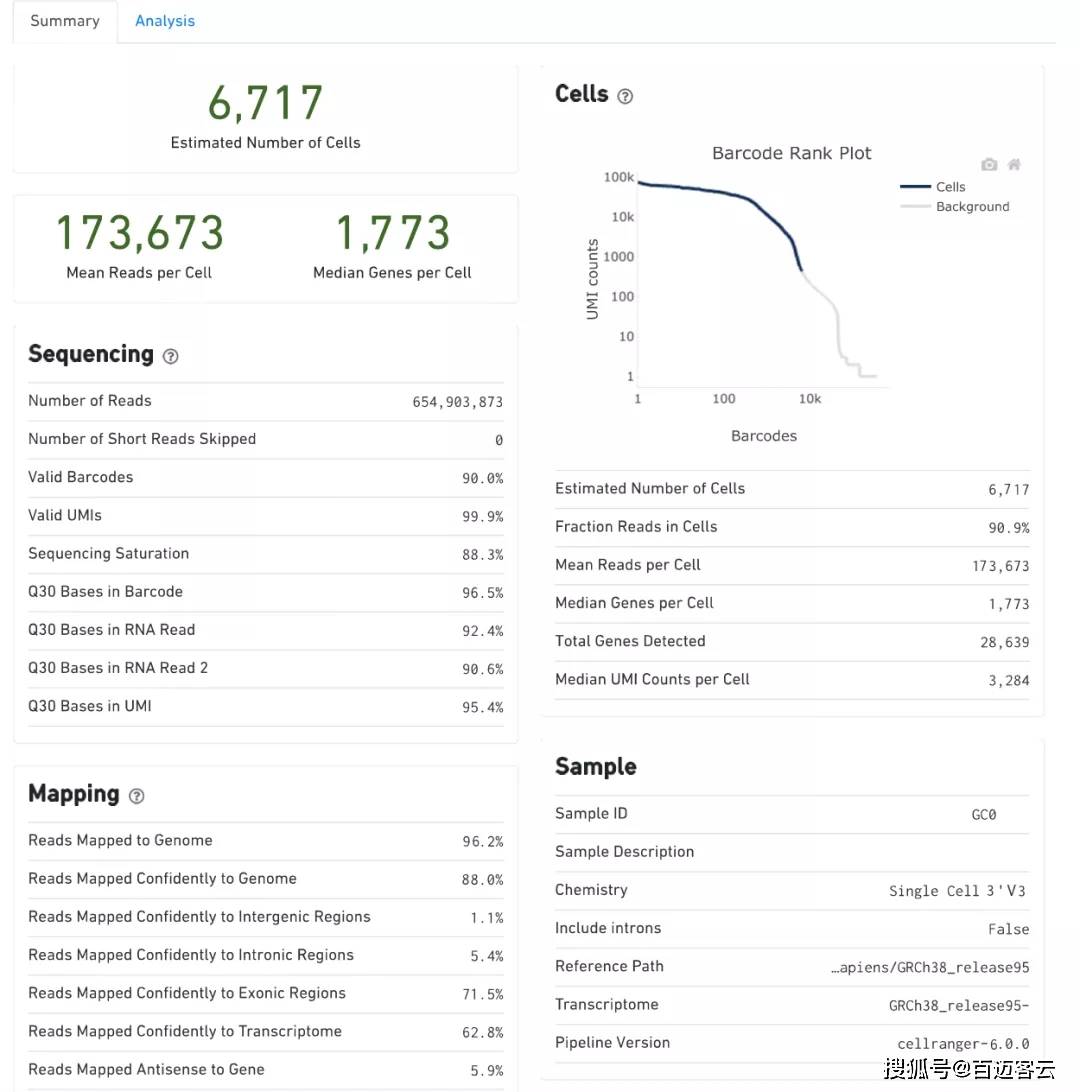 图3 summary结果
图3 summary结果
二、数据过滤
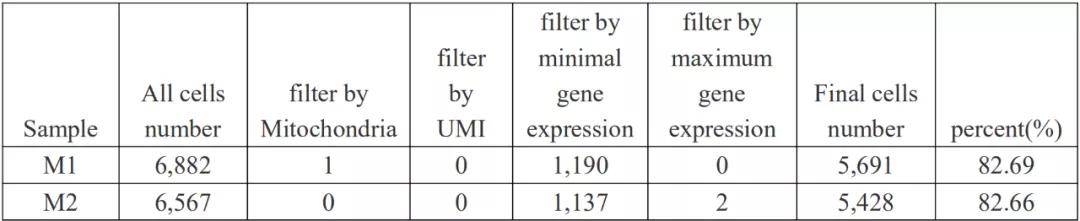
我们在根据CellRanger分析得到细胞基因表达矩阵结果后,使用Seurat软件做进一步分析。首先需要对数据进一步过滤,主要指标包括:细胞表达的基因数目、细胞的UMI总数、线粒体基因占比。
细胞的最低基因数目一般在200-500之间,因此过滤参数设置为:
(1) 基因表达的细胞数:>= 10;
(2) 线粒体含量(%):<= 20;
(3) UMI数目:>= 100;
(4) 基因数目:500 ~ 7000。
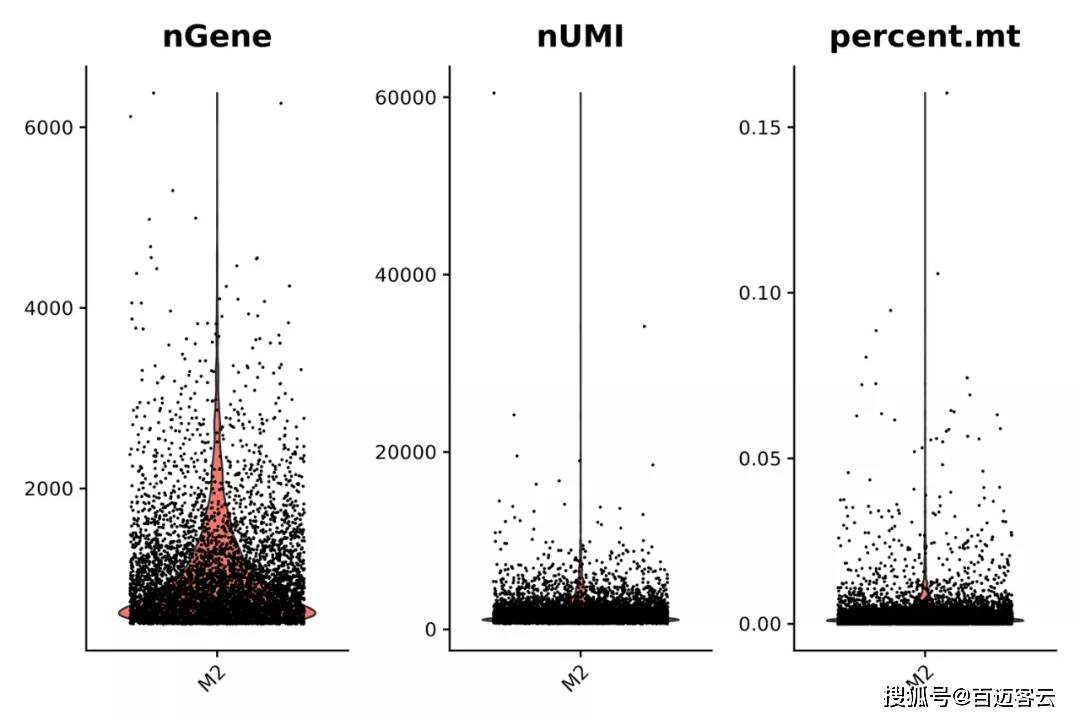 图4数据过滤后
图4数据过滤后
通过上述过滤标准统计细胞过滤的数目情况,最终后续分析基于高质量的细胞进行细胞聚类、差异分析、功能富集分析、细胞注释等。
三、细胞聚类分析
筛选得到有效细胞后,会对数据进一步做标准化处理。默认标准化方法为LogNormalize,Seurat内置的FindVariableFeatures()函数,用来计算每一个基因的均值和方差,默认选择高变的2000个基因用于下游分析。ScaleData()函数对数据进行线性转换即归一下处理,使得每一个基因在所有cell中的表达均值为0,方差为1。
目前常用的降维算法包括:PCA(Principal Component Analysis)、t-SNE(t-Distributed Stochastic Neighbor Embedding)、UMAP(Uniform Manifold Approximation and Projection)。
T-SNE分析主要是对细胞进行聚类,根据细胞的表达模式进行分群,一般认为同一种细胞类型的细胞基因表达模式相似。细胞分群的数目由resolution参数决定,百迈客resolution参数默认设置为0.2,根据可视化的结果可以调节resolution参数来改变细胞cluster的数目。当细胞cluster数目偏少,cluster中细胞数目偏多,可以将resolution参数调大,最大到1.0。
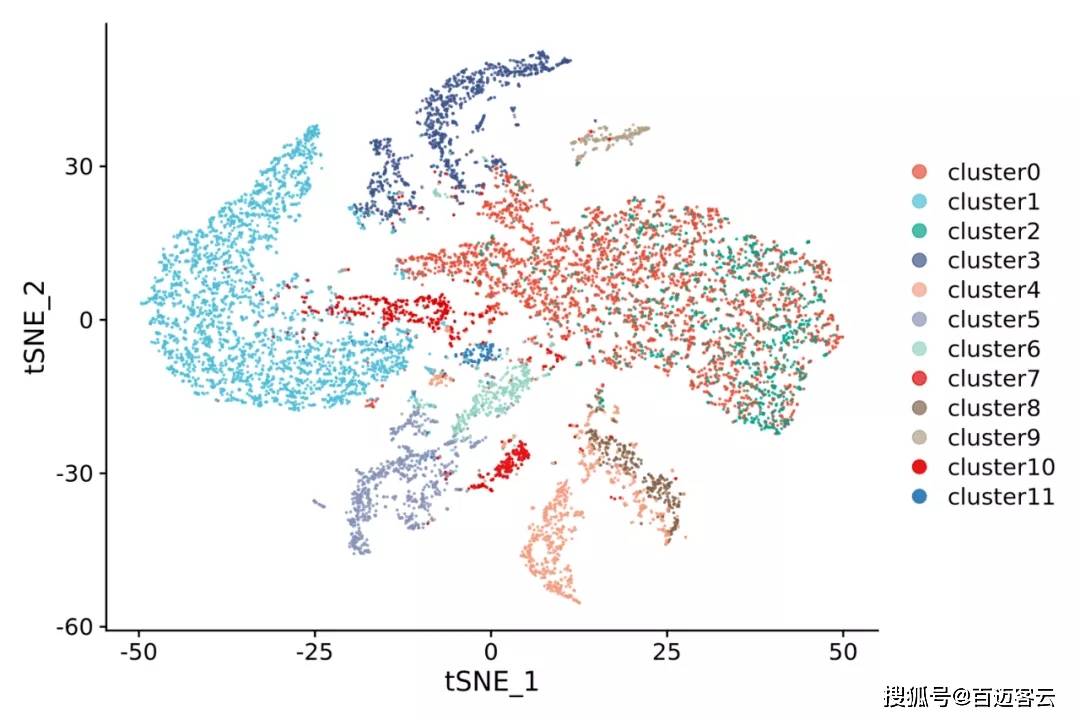 图5细胞聚类的t-SNE图
图5细胞聚类的t-SNE图
四、差异表达基因及功能分析
上面我们对细胞进行了分群,我就需要对不同的细胞类型的差异基因及功能属性的差异进行分析。并基于细胞cluster之间的差异对细胞进行注释。
采用Seurat的wilcox秩和检验对不同类的细胞筛选差异表达基因。将某一类细胞的基因与其他所有类的基因做比较,默认筛选至少在一个比较组中超过25%的细胞中表达的基因进行差异比较分析。差异筛选标准为 Fold Change≥ 2 且Thred < 0.1(parameter is: FDR)。
针对各个cluster的差异基因我们也进行了功能富集分析,用于更好的判断不同细胞类群行使的作用。百迈客除了提供了常规的GO和KEGG数据库外,还提供了Reactome数据的注释。
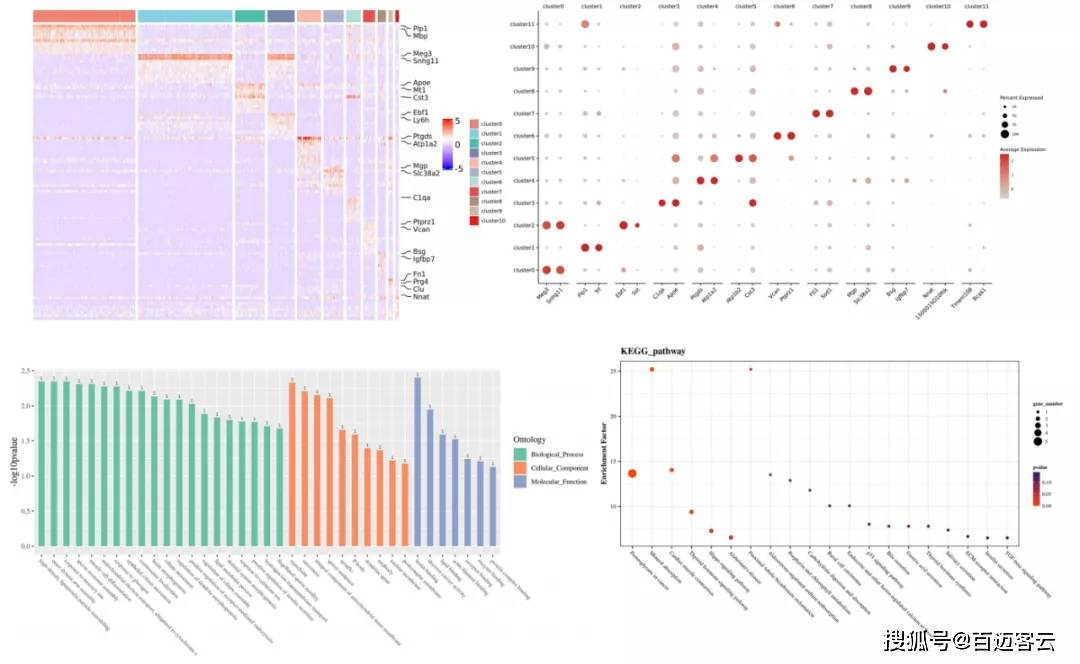 图6 各cluster差异基因热图、气泡图、富集柱状图和富集气泡图
图6 各cluster差异基因热图、气泡图、富集柱状图和富集气泡图
五、转录组因子结合位点预测
转录组因子(TF)能够与基因启动子区域中顺式作用元件发生特异性相互作用的DNA结合蛋白,通过它们之间以及与其他相关蛋白之间的相互作用,激活或抑制某些基因的转录。
百迈客使用TFBStools对差异基因的启动子区域上的 TFBS 进行了预测,参考的转录因子motif数据库是JASPAR数据库(http://jaspar.genereg.net/)。差异基因上游启动子区域有转录因子的结合,可能是导致该基因差异表达的原因。
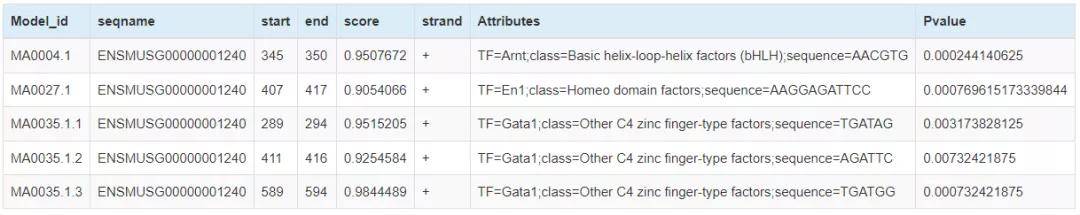 图7转录因子结合位点预测结果
图7转录因子结合位点预测结果
六、多样品合并分析
科学研究时一般会涉及到很多样品,需要对多个样本的数据进行合并以及数据的均一化。将样品数据进行统一标准才能更好的进行后续的细胞类型的注释以及组件的差异分析。
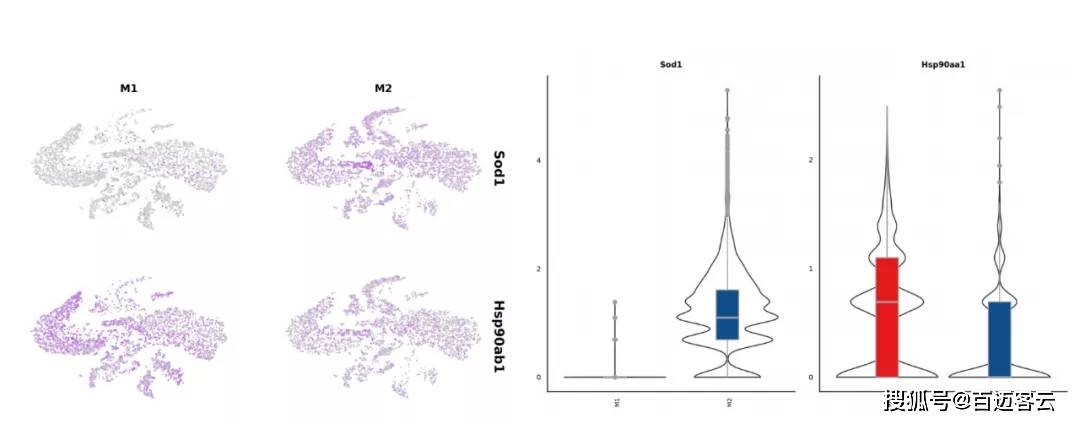 图8 marker基因t-NSE图和小提琴图
图8 marker基因t-NSE图和小提琴图
七、细胞注释分析
单细胞注释主要包含两种思路,一种是将未知细胞类型的细胞数据跟已知类型的细胞数据进行相关性分析,从而对未知的表达数据进行细胞类型鉴定。另一种方法是根据marker gene进行鉴定,将聚类得到的亚群的marker 基因跟数据库中已知的细胞类型的marker基因进行比较。SingleR是基于斯皮尔曼相关性,对scRNA-seq数据实现自动化注释的一个软件。
百迈客使用 SingleR 对细胞类群进行自动注释(只针对人、小鼠)。内置5个人数据库和2个小鼠数据库,主要包括HumanPrimaryCellAtlasData和MouseRNAseqData。当然除了SingleR软件外,百迈客还有SCINA、scCATCH等软件可以进行细胞注释分析。细胞注释详见微信稿:https://mp.weixin.qq.com/s/W3lKqdZyeejCqs_atPV3LA
 图9 SingleR内置7个数据库
图9 SingleR内置7个数据库
细胞类型鉴定的过程即复杂又繁琐,自动注释只能提供一个参考。然后根据实际情况参考相关的marker基因进行手段校正。2019年1月CellMarker发表于Nucleic Acids Research,CellMarker数据库(http://biocc.hrbmu.edu.cn/CellMarker/)收录了158种组织/亚组织的467种人细胞类型, 81种组织/亚组织的389种鼠细胞类型。数据主要来源于文献和数据库,包括单细胞测序数据和生物实验数据。
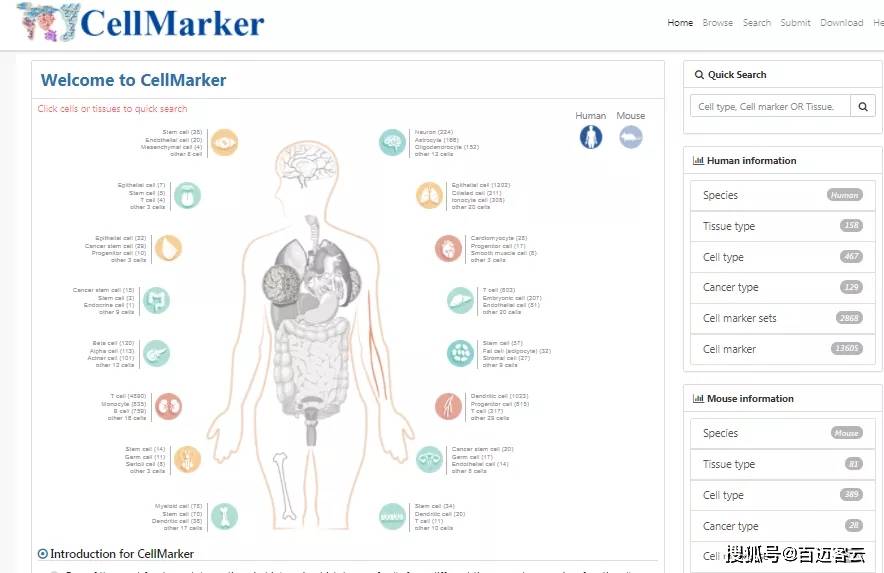 图10 CellMarker数据库
图10 CellMarker数据库
当我们的样品分组涉及到不同处理时,需要比较同一种细胞类型在不同处理组中的占比情况以及基因的差异和功能的变化。
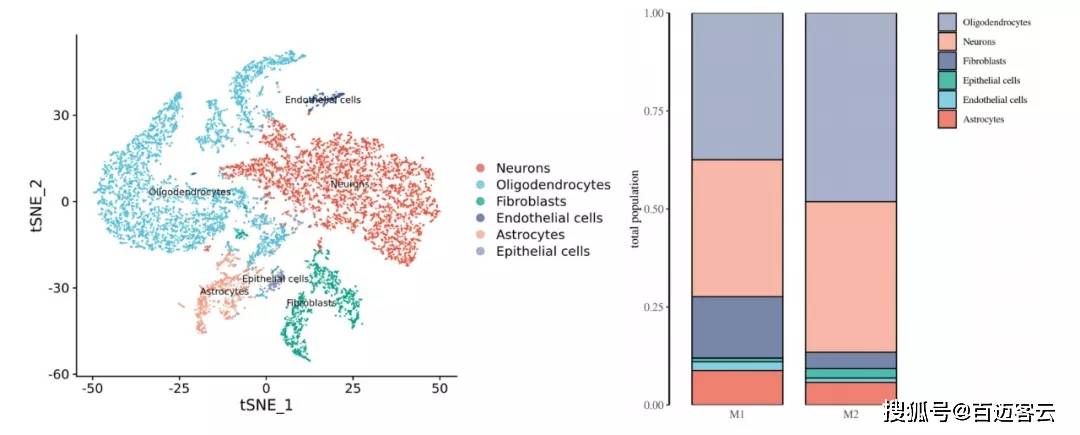 图9细胞类型t-NSE图和分布统计图
图9细胞类型t-NSE图和分布统计图
上述单细胞分析流程需要的配置及生信功底要求很高,对于没有生信基础、不会敲代码、不会跑流程、没有服务器等硬件设备的老师来说,可以使用百迈客单细胞转录组(scRNA-seq)云分析平台,轻松get单细胞转录组初级分析。
 图10 百迈客单细胞转录组云平台
图10 百迈客单细胞转录组云平台
单细胞转录组研究思路
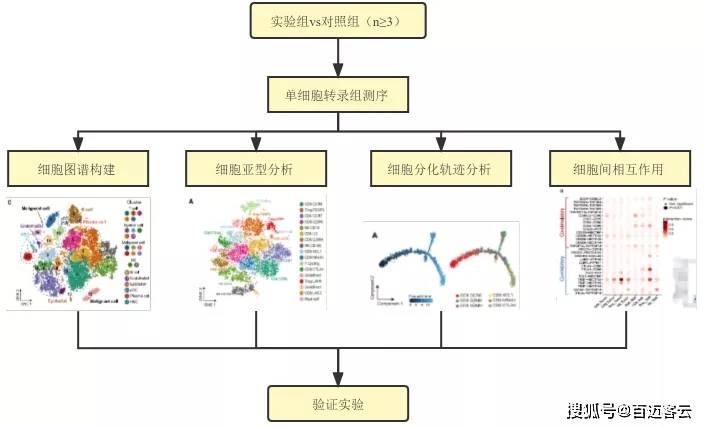 图11单细胞研究思路
图11单细胞研究思路
参考文献
【1】Single-cell RNA sequencing shows the immunosuppressive landscape and tumor heterogeneity of HBV-associated hepatocellular carcinoma
【2】Single-cell landscape of the ecosystem in early relapse hepatocellular carcinoma
【3】https://www.10xgenomics.com/products
【4】http://biocc.hrbmu.edu.cn/CellMarker/- 本文固定链接: https://maimengkong.com/zu/880.html
- 转载请注明: : 萌小白 2022年4月30日 于 卖萌控的博客 发表
- 百度已收录
