
这两年随着测序成本的下降和转录组研究的日渐火热,RNA-seq俨然已经成为了分子生物学课题组推进项目的首选方向。在我们接触的转录组项目中,有些老师对项目分析结果存在或多或少不清楚或有疑惑的地方。那么春天来了,花儿开了,今天福利也到了,我们特意将转录组项目中常见的一些问题进行了汇总,各位老师可以按需自取哈。
1.如何判定生物学重复一致性的高低?生物学重复统计方法及公式
答:(1)皮尔逊相关系数r可以作为生物学重复相关性的评估指标,理想的生物学重复试验r2≧0.92。考虑到个体差异、取材环境、时间以及人员操作熟练程度等因素对测序数据的影响,一般r2≧0.8为可接受范围。
(2)Pearson(皮尔逊)相关系数:皮尔逊相关也称为积差相关(或积矩相关)是英国统计学家皮尔逊于20世纪提出的一种计算直线相关的方法。
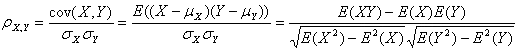
2.DEG基因用Transcripts还是Unigenes?
答:DEG基因用的是Unigene。
3.transcript-id代表什么意思?为什么有的基因有多个transcript-id?
答:基因转录本id;因为可变剪切的缘故,一个基因可能有多个转录本。
4.在miRNA鉴定中,可能成为miRNA的reads是怎样计算的?哪些条件会影响到mrd值?micro RNA在不同组织有异构体的存在,是如何处理的?
答:与 Rfam, miRbase, RepBase和 Exon\Intro 序列库进行比对,获得 sRNA 注释信息,以此作为预测新的 miRNA 的基础。
miRNA的鉴定是利用miRDeep2软件进行已知及新(保守及非保守)的miRNA鉴定。miDeep2会在reads比对到基因组上的位置两端分别延伸75、15bp进行结构预测,此软件认为极可能与可能是miRNA的根据是通过mrd值来区分的,mrd>-10为可能,mrd>0为极可能;
影响mrd值的有reads在基因组上的分布和碱基结合的自由能等;
5.对于有生物学重复的项目,怎样计算差异基因?
答:两两比对使用的是R的EBseq包, 是基于负二项分布检验的方式对reads数进行差异显著性检验,重复间的比对使用的是R的DEseq包,是基于分层贝叶斯模型的原理对组合内样品进行分析。
6.外显子,内含子及基因间区各自的比例如何评估建库情况?
答:理论上,来自成熟mRNA的reads应该比对到外显子区。但是,由于基因组注释水平、可变剪切导致的内含子序列保存,以及很多RNA(比如lncRNA)就来自基因间区和内含子,因此有比对到内含子和基因间区的reads。受物种等的影响外显子所占比例不同,一般情况下外显子区域所占比例超过70%即比较理想。
7.影响组装Contig结果的因素?
答:a.物种的特异性;b.测序质量;c.测序的数据量;d.SNP的杂合率;e.组装参数的选择。
(1)、在不考虑物种特异性和测序质量的情况下,测序的数据量越大,SNP的杂合率越高,得到的短片段Contig的数目就越多。根据Trinity组装Contig的策略,将Reads构建K-mer库,选取频数最高的K-mer,按照k-1的overlap进行延伸,用于延伸的K-mer全部从库中清掉,因此测到的reads越多,SNP的杂合率越高,延伸完后的短片段就越多。
(2)、对于组装参数的选择,是用于过滤低频数K-mer,选择的参数不同,过滤掉的K-mer数目不同,如果过滤掉的越多,那么留下的短片段的Contig就会少。所以即使用同一个软件(Trinity)进行组装,如果不知道组装参数的时候,对于组装结果没有很大的可比性。
(3)、组装结果的好坏最主要的还是看Unigene的组装数据,包括组装出的数目和N50。一般来说,组装出的Unigene的数目在一个合理范围内(比如10W以内),N50越大,组装的结果越好。
8.转录组测序Contig 与transcript的区别?
答:转录组测序的原始数据包含了很多的reads,通过序列的拼接,具有重叠区的reads会被组装成更大的片段,称之为contig。将reads比对回contig,通过paired-end reads能确定来自同一转录本的不同contig 以及这些contig之间的距离,将这些contig连在一起,最后得到两端不能再延长的序列,称之为Unigene。Transcript即转录本。
9.不同ID号代表的基因相同吗?不同ID号功能注释相同的,为什么?
答:不同的ID可以认为是代表不同的基因。不同的基因注释的功能相同,原因有:一是有些长的基因没有组装出完整的序列,而是分成了多个小片段,这种情况去进行注释的话会注释到同一个功能蛋白;二是基因的核酸序列不同,但是蛋白序列具有一定的相似性或者具有相似的功能区域,这些基因在比对注释用的蛋白序列时,会注释到相同的功能。
10.多个Unigene注释一样,序列长度不同,相似性较低,为什么?
答:1)首先某一基因可能比较长,但无参考基因组装出的片段即Unigene很难组装得到全长,得到的是这个基因上的大小不等的片段,在进行比对的时候就会比对到同一个基因上,因此他们的注释信息一致;
2)从序列来看Unigene基因的序列相似度不高,但是因为比对的是蛋白,所以可能他们的蛋白相似度会比较高,因此会注释到同一基因上。
11.transcript_id、gene_id、length、effective_length、expected_count、TPM、FPKM、IsoPct这几个字段的意思?
答:一个Unigene可能对应多个转录本。Transcript id:为组装转录本编号;gene_id:Unigene编号;length:Unigene的长度;effective_length:各个转录本的平均长度;TPM:Transcripts per million,公式为:Unigene 的reads数×10^6/总reads数;FPKM即RPKM(双端Reads数目/(比对到转录本上的片段总数*转录本长度));IsoPct:某一个转录本的表达量占相应的组装原件表达量的百分比。
12.同一ID下有多条序列,想得到此序列的核苷酸信息应选哪一条?
答:同一个ID号下面好几条序列,这个应该是组装过程中装出来的转录本序列,来自同一个Component(具体见Trinity组装的第二步),其ID前缀相同,后面跟着seq+数字的编号。Trinity软件认为这些转录本来源于同一个基因,因此,选取其中最长的那个转录本的序列作为该基因的序列。
13.生物云转录组APP上的差异筛选阈值采用的是哪种方法?p值与FDR值的区别是?
答:生物云转录组APP在差异表达分析过程中采用了公认有效的Benjamini-Hochberg方法对原有假设检验得到的显著性p值(p-value)进行校正,并最终采用校正后的p值,即FDR(False Discovery Rate)作为差异表达基因筛选的关键指标,以降低对大量基因的表达值进行独立的统计假设检验带来的假阳性。p值与FDR之间没有单纯的换算公式,是在linux操作系统下,运用R语言编写的程序完成的fisher精确检验,在筛选过程中,默认将FDR<0.01且差异倍数(Fold Change)≥2作为筛选标准。
14.生物云转录组在分析差异基因时,对于表达量为0的,如何计算差异倍数?
答:差异基因分析软件EBseq在分析表达量为0的基因的差异倍数时,会采用贝叶斯估计给出一个估计值,然后使用这个估计值计算差异倍数。由于计算估计值时综合考虑多项因素,因此不同基因间FPKM和FC不具有一致性。
15.如何定义的已知micRNA、保守的micRNA以及新预测的micRNA?
答:已知micRNA指的是序列在miRBase数据库中百分百的比对到该物种的序列上,如果在该物种上没有比对上但比对上了数据库中的其他物种上我们称之为保守的micRNA;新预测的micRNA:通过miRDeep2软件进行预测,有一定的read能够比对到基因组上,并且比对位置的序列可以形成发卡结构,那么就会作为新预测的miRNA。
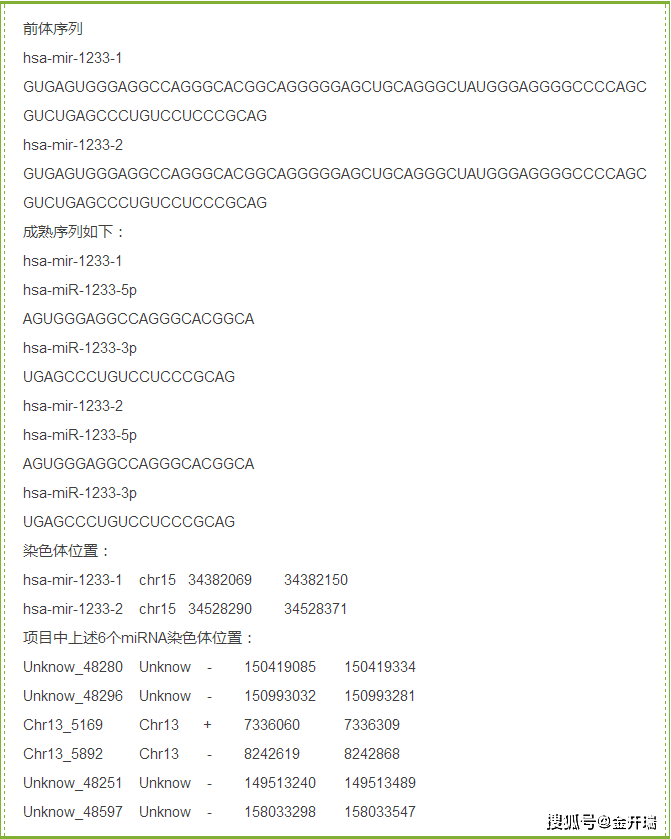
16.分析时发现不同的名,但是他们的前体序列和成熟序列都一样,表达量在各个样品中也相同,为什么?
答:这个是由于在染色体上的位置不同导致的,可以参考miRBase数据库中的 hsa-mir-1233-1 和 hsa-mir-1233-2 这两个 ID, 它们对应的前体序列,3' 和 5' 成熟序列均相同,但在基因组上的位置不同,软件将它们区别成两个不同的小RNA,又因为它们的序列一致,所以比对上的reads是一样的,表达量因此一样。具体见下:
17.测序得到的lncRNA,如何知道哪些是已知的?哪些是未知的?
答:目前长链分析结果中如果分析的物种是比较常见的物种比如人、大鼠、小鼠,这些物种具有比较完整的已知lncRNA数据库,这种情况:
(1)通过确切的位置关系(位置相交则认为相同)对预测出来的那些lncRNA鉴定其是否为已知;
(2)根据fa序列进行比对,对预测出的lncRNA序列与数据库中已知的lncRNA序列比对,达到一定比对值的会认为该预测长链是已知的长链。
注:NONCODE DB中包含的物种主要是动物方面的,包括:人、小鼠、大鼠、奶牛、鸡、果蝇、斑马鱼、线虫、酵母、拟南芥、黑猩猩、大猩猩、恒河猴、复鼠、鸭嘴兽、猩猩
18.转录组测序之后,用QPCR进行验证,但验证的基因表达趋势与测序结果中不一致,这是什么原因?如何解决呢?
答:首先,我们需要确定检验的样品是否是同一批次,验证样品的上下调关系是否与测序结果中的一致(这个需要根据测序公司具体的分析结果,比如某个基因的FC值对应的样品写的是T01 vs T02 ,那么T01就是对照组、T02是实验组),若样品不为同一批次或其上下调关系颠倒了,则势必会导致验证基因表达趋势不一致的情况。
其次,我们需要查看验证基因的表达量、样品和实验用的引物是否被污染,若验证基因表达量过低,则有可能导致差异不显著,若样品或实验用的引物被污染则后续结果可能也不会准确,所以我们尽量不要挑选表达量太低的基因,同时,需要保证样品和实验引物没有被污染。
当以上所有情况都不存在,且结果依然不一致,这时我们需要检查QPCR结果是否正确。如果仅一个基因验证结果不一致,则不足以说明测序或者验证有问题,但当我们选择了15个基因甚至更多时,结果依然不一致时,那么我们可能需要分析测序数据的结果是否正确,同时检查结果预期是否正确。
19.从NCBI上下载的数据都是SAR格式的,如何转化成FASTQ格式?
答使用软件sra2fastq进行转换。
20.Trans:转录组插入片段检测为什么有小峰?
答:这种情况只有出现在有参转录组的插入片段检测处为正常,其它的分析出现此情况都是不正常现象。有参中出现是因为测序reads与参考基因组比对造成的,转录组的测序数据都是基因组上外显子区域,但是在比对时,双端reads是比对到了基因组上两个相邻外显子的区域,那么在检测插入片段时,就会出现比实际序列长的现象,因此在有参转录组的插入片段检测时,出现在主峰右边的小峰为正常,出现在其左边或者其他分析,都是不正常的。- 本文固定链接: https://maimengkong.com/zu/873.html
- 转载请注明: : 萌小白 2022年4月22日 于 卖萌控的博客 发表
- 百度已收录
