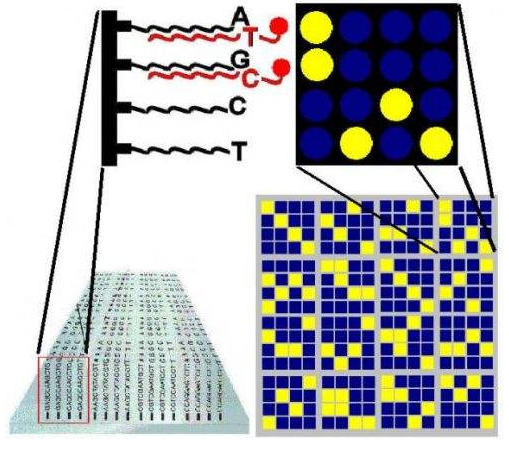
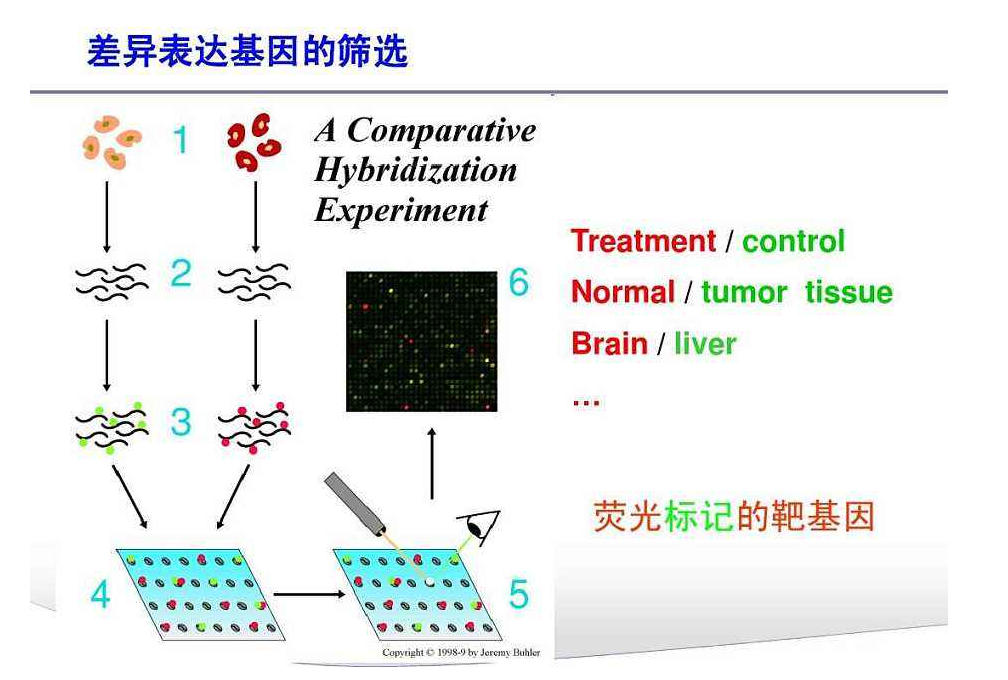
基因芯片(Gene chip)(又称DNA芯片、生物芯片)最初是80年代中期提出的。它是由大量DNA或寡核苷酸探针密集排列所形成的探针阵列,其工作的基本原理是通过碱基互补配对检测生物信息。即通过与一组已知序列的核酸探针杂交进行核酸序列测定的方法,在一块基片表面固定了序列已知的靶核苷酸的探针。当溶液中带有荧光标记的核酸序列TATGCAATCTAG,与基因芯片上对应位置的核酸探针产生互补匹配时,通过确定荧光强度最强的探针位置,检测到一组序列完全互补的探针序列。据此可重组出靶核酸的序列。
目前已有许多数据库,包括NCBI的GEO数据库(https://www.ncbi.nlm.nih.gov/geo/),ArrayExpress数据库(https://www.ebi.ac.uk/arrayexpress/),和TCGA数据库(https://cancergenome.nih.gov/)等等,记录和储存着大量芯片相关的数据,其中GEO数据库是目前最大最全的数据库,可供科研人员查询和下载相关数据。
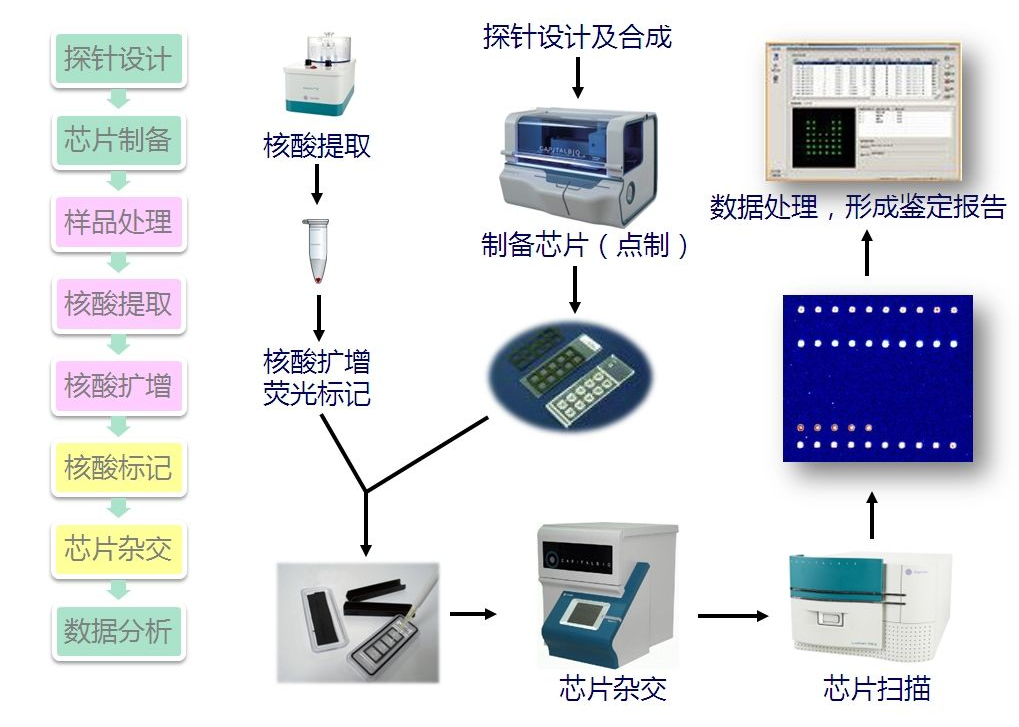
下面和大家分享一下基因芯片数据的预处理方法。
1)分析前需要对数据进行背景信号处理:背景处理即过滤芯片杂交信号中属于非特异性的背景噪音部分。一般以图像处理软件对芯片划格后,每个杂交点周围区域各像素吸光度的平均值作为背景,但此法存在芯片不同区域背景扣减不均匀的缺点。也可利用芯片最低信号强度的点(代表非特异性的样本与探针结合值)或综合整个芯片非杂交点背景所得的平均吸光值做为背景。
背景处理之后,我们可以将芯片数据放入一个矩阵中:

其中,各字母的意义如下:
N:条件数;
G:基因数目(一般情况下,G>>N);
行向量mi=(mi1,mi2,…,miN)表示基因i在N个条件下的表达水平(这里指绝对表达水平,亦即荧光强度值);
列向量mj=(m1j,m2j,…,mGj)表示在第j个条件下各基因的表达水平(即一张芯片的数据);
元素mij表示第基因i在第j个条件下(绝对)基因表达数据。m可以是R(红色,Cy5,代表样品组)。也可以是G(绿色,Cy3,代表对照组)。
2)芯片数据清理:经过背景校正后的芯片数据中可能会产生负值,还有一些单个异常大(或小)的峰(谷)信号(随机噪声)。对于负值和噪声信号,通常的处理方法就是将其去除,常见数据经验型舍弃方法有:A.标准值或奇异值舍弃法;B.变异系数法;前景值<200;前景值-平均数/前景值-中位数<80%等等。然而,数据的缺失对后续的统计分析(尤其是层式聚类和主成分分析)有致命的影响。Affymetrix公司的芯片分析系统会直接将负值修正为一个固定值。
缺失值得处理方法:对数据的删除,通常是删去所在的列向量或行向量。一个比较常用的做法是,事先定义个阈值M。若行(列)向量中的缺失数据量达到阈值M,则删去该向量。若未达到M,有两种方法处理,一是以0或者用基因表达谱中的平均值或中值代替,另一个是分析基因表达谱的模式,从中得到相邻数据点之间的关系,据此利用相邻数据点估算得到缺失值(类似于插值)。填补缺失值(k临近法):利用与待补缺基因距离最近的k个临近基因的表达值来预测待填补基因的表达值。
3)提取芯片数据的表达值:由于芯片数据的小样本和大变量的特点,导致数据分布呈偏态、标准差大。对数转换能使上调、下调的基因连续分布在0的周围,更加符合正态分布,同时对数转换使荧光信号强度的标准差减少,利于进一步的数据分析。
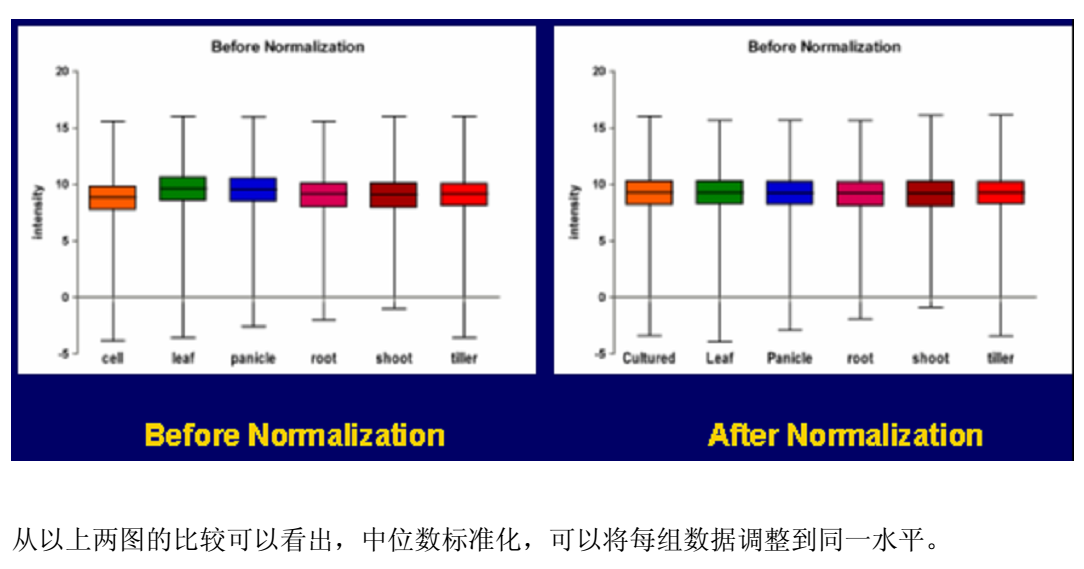
4)芯片数据的归一化:经过背景处理和数据清洗处理后的修正值反映了基因表达的水平。然而在芯片试验中,各个芯片的绝对光密度值是不一样的,在比较各个试验结果之前必需将其归一化(normalization,也称作标准化)。数据的归一化目的是调整由于基因芯片技术引起的误差,不是调整生物RNA 样本的差异。在同一块芯片上杂交的、由不同荧光分子标记的两个样品间的数据,也需归一化。常用的方法是平均数、中位数标准化(mean or median normalization):将各组实验的数据的log ratio中位数或平均数调整在同一水平。中位数标准化:将每个芯片上的数值减去各自芯片上log Ratio值的中位数,使得所有芯片的log Ratio值中位数就变成了0,从而不同芯片间log Raito具有可比性。
5) 差异基因表达分析: 经过预处理,探针水平数据转变为基因表达数据。为了便于应用一些统计和数学术语,基因表达数据仍采用矩阵形式。
A.芯片数据的差异分析主要包括三种方法:
1. 倍数分析方法:倍数变换fold change,单纯的case与control组表达值相比较,对没有重复实验样本的芯片数据,或者双通道数据采用这种方法。
2. 参数法分析(t检验):当t超过根据可信度选择的标准时, 比较的两样本被认为存在着差异。但小样本基因芯片实验会导致不可信的变异估计,此时采用调节性T检验。
3. 非参数分析:由于微阵列数据存在“噪声”干扰而且不满足正态分布假设,用t检验有风险。非参数检验并不要求数据满足特殊分布的假设,所以可使用非参数方法对变量进行筛选。如经验贝叶斯法、芯片显著性分析SAM法。
B. 芯片数据的差异分析的常用软件包括:
1. Limma:它是一个功能比较全的包,既含有cDNA芯片的RAW data输入、前处理(归一化)功能,同时也有差异化基因分析的“线性”算法(limma: Linear Models for Microarray Data),特别是对于“多因素实验(multifactor designed experiment)”。limma包的可扩展性非常强,单通道(one channel)或者双通道(tow channel)数据都可以分析差异基因,甚至也包括了定量PCR和RNA-seq。
2. DESeq2和EdgeR包: 都可用于做基因差异表达分析,主要也是用于RNA-Seq数据,同样也可以处理类似的ChIP-Seq,shRNA以及质谱数据。这两个都属于R包,其相同点在于都是对count data数据进行处理,都是基于负二项分布模型。
3. GFOLD软件:对于有生物学重复的数据(一般的转录组数据都会有生物学重复),我们一般采用一个叫edgeR和DEseq的R包。但如果预先测了一批数据没有重复的数据进行一个预分析。这时候edgeR依然可以用,不过需要认为指定一个dispersion值,这样的不同的人就可以有不同的结果,在查阅了很多资料之后呢,大家一致认为没有重复的转录组数据应该用GFOLD软件。
转自生信草堂公众号- 本文固定链接: https://maimengkong.com/kyjc/874.html
- 转载请注明: : 萌小白 2022年4月23日 于 卖萌控的博客 发表
- 百度已收录
