来源:生物通
新一代测序技术在爆炸式发展的同时,也衍生出许多其他技术创新。RNA深度测序(RNA-Seq)就是其中之一,这项技术使我们对细胞发育及其调控机制的理解,达到了前所未有的深度和广度。尽管研究细胞RNA并不是什么新鲜事,但RNA-Seq的出现大大拓展了转录组研究的规模,取得了累累硕果,这些是传统技术难以企及的。
RNA-seq可以获得相当惊人的数据量,而这恰恰是一柄双刃剑。丰富的数据量蕴含着大量的宝贵信息,但这样的数据需要复杂的生物信息学分析,才能从中提取到有意义的结果。正因如此,数据分析可以说是RNA-seq的重中之重。
RNA-seq有非常广泛的应用,但没有哪个分析软件是万能的。科学家们一般会根据自己的研究对象和研究目标,采用不同的数据分析策略。现在人们已经发表了大量的RNA-seq和数据分析方案,对于刚入门的新手来说难免有些无所适从。
佛罗里达大学、加州大学Irvine分校等单位的研究人员在一月二十六日的Genome Biology杂志上发表文章,概述了RNA-seq生物信息学分析的现行标准和现有资源,为人们提供了一份带有注释的RNA-seq数据分析指南。这将成为开展RNA-seq研究的宝贵参考资料。
这份指南覆盖了RNA-seq数据分析的所有主要步骤,比如质量控制、读段比对、基因和转录本定量、差异性基因表达、功能分析、基因融合检测、eQTL图谱分析等等。研究人员绘制的RNA-seq分析通用路线图(标准Illumina测序),将主要分析步骤分为前期分析、核心分析和高级分析三类。前期预处理包括实验设计、测序设计和质量控制。核心分析包括转录组图谱分析、差异基因表达和功能分析。高级分析包括可视化、其他RNA-seq技术和数据整合。
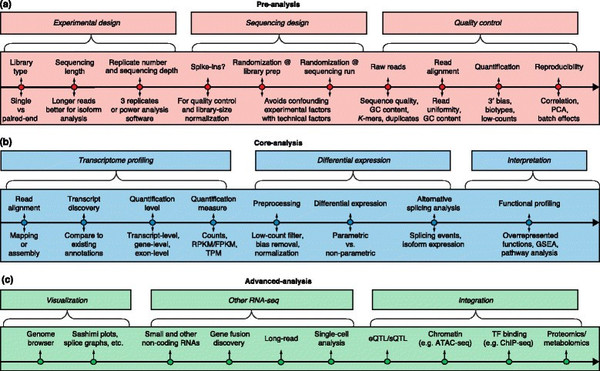
研究人员在文章中探讨了每个步骤所面临的挑战,也评估了一些数据处理方法的潜力和局限。此外,他们还介绍了RNA-seq数据与其他数据类型的整合。这种数据整合可以将基因表达调控与分子生理学和功能基因组学关联起来,如今越来越受到研究者的欢迎。
这篇文章在结尾处介绍了一些为转录组领域带来改变的新技术,特别是单细胞RNA-seq和长读取测序技术带来的机遇和挑战。
2015年年初,RNA-Seq的数据分析方法如雨后春笋般涌现。三月份,Nature集团旗下刊物发表了三篇介绍RNA-Seq数据分析新方法的文章,一篇发表在《Nature Methods》上,另外两篇发表在《Nature Biotechnology》上。这三篇文章有一位共同的作者,那就是约翰霍普金斯大学计算生物学中心的Steven Salzberg,生物信息学和计算生物学领域的杰出科学家。Salzberg通过这些文章中分别介绍了三种新工具:HISAT、StringTie和Ballgown。这些工具可以取代之前开发的早期工具,为RNA-Seq提供了全新的数据分析方法,从原始数据读取到差异表达分析。
RNA测序究竟有多可靠呢?由美国FDA牵头的测序质量控制(SEQC)项目对RNA测序的准确性、可重现性和信息含量进行了综合性评估。其初步调查结果发表在2014年09月的Nature Biotechnology杂志上,石乐明教授是这篇文章的通讯作者之一。研究人员用RNA参照样本在全球多个实验室的Illumina HiSeq、Life Technologies SOLiD、Roche 454平台上进行检测,主要评估RNA测序在接头区域和差异性表达谱中的表现,并将其与芯片和定量PCR(qPCR)进行比较。研究表明,数据分析的算法会对RNA测序产生很大影响,不同算法生成的转录本数据存在很大差异。
前几天,浙江大学和哈佛大学的研究人员在Cell Reports杂志上发表了一项单细胞mRNA-seq研究。基因表达变异是小鼠胚胎干细胞(ESC)的一个重要特征,但人们一直不清楚这背后的具体原因。研究人员通过分析小鼠胚胎干细胞发现,这些细胞表现出的异质性是血清培养造成的。他们在其中鉴定了高度变异的基因簇,以及独特的染色质状态。研究显示,双价基因(bivalent gene)更容易出现表达变异。进一步研究表明,无血清培养可以减少小鼠ESC的异质性和转录组变异。这意味着,细胞内的网络变异大多是细胞外的培养环境造成的。
End
- 本文固定链接: https://maimengkong.com/zu/1117.html
- 转载请注明: : 萌小白 2022年7月10日 于 卖萌控的博客 发表
- 百度已收录
