Seurat对象剖析以及Seurat Command List
前景回顾
首次揭秘!不做实验也能发10+SCI,CNS级别空间转录组套路全解析(附超详细代码!)
过关神助!99%审稿人必问,多数据集联合分析,你注意到这点了吗?
太猛了!万字长文单细胞分析全流程讲解,看完就能发文章!建议收藏!(附代码)
秀儿!10+生信分析最大的难点在这里!30多种方法怎么选?今天帮你解决!
图好看易上手!没有比它更适合小白入手的单细胞分析了!老实讲,这操作很sao!
Seuart对象
#让我们回顾一下标准的流程,然后我们来一步步解析Seurat对象 pbmc.counts <- Read10X(data.dir = "~/Downloads/pbmc3k/filtered_gene_bc_matrices/hg19/") pbmc <- CreateSeuratObject(counts = pbmc.counts) pbmc <- NormalizeData(object = pbmc) pbmc <- FindVariableFeatures(object = pbmc) pbmc <- ScaleData(object = pbmc) pbmc <- RunPCA(object = pbmc) pbmc <- FindNeighbors(object = pbmc) pbmc <- FindClusters(object = pbmc) pbmc <- RunTSNE(object = pbmc) DimPlot(object = pbmc, reduction = "tsne")
晨曦解读
然后接下来我们一步步来解析Seurat对象,只有对Seurat对象中的数据存储有一个最基本的了解,我们才可以开始学习Seuart Command List~
#读取数据 library(Seurat) pbmc <- Read10X(data.dir = "hg19/")
晨曦解读
这三个文件代表一个数据集,分别是细胞ID文件(barcode.tsv)、基因信息文件(gene.tsv)以及表达量文件(matrix.mtx),反正具体来说细究这个倒是没有太多的意义,因为后续我们很少会用到这类数据,基本上就是改好文件名读取进来即可~
#因为我们前面介绍过,Seurat对象是一个类似于不断补充的对象,并不会因为赋值而替换,所以这里我们走完标准流程,然后针对标准流程后的Seurat对象进行解析 #正好我这里有一个Rdata,所以这里我直接加载进来了 scRNA <- readRDS("~/scRNA.rds") scRNA@assays[["RNA"]]@counts#原始数据,就是我们读取那三个文件后获得的原始数据 scRNA@assays[["RNA"]]@data#经过标准化的数据 scRNA@assays[["RNA"]]@scale.data#经过标准化后执行高可变基因筛选并且归一化后的数据
晨曦解读
也可以戏称为基础三数据,也就是原始、标准、高可变这三个维度的数据~
然后assays这个分区内就没有什么感兴趣的数据了,然后我们往下看
scRNA@meta.data[["orig.ident"]]#样品名称 scRNA@meta.data[["nCount_RNA"]]#每个细胞中RNA的含量 scRNA@meta.data[["nFeature_RNA"]]#每个细胞中基因的含量 scRNA@meta.data[["percent.mt"]]#每个细胞线粒体基因的占比百分比 scRNA@active.assay#scRNA-seq的类型

晨曦解读
meta.data这个分区主要存放着一些质控的信息~
scRNA@active.ident#降维聚类后的信息,格式为细胞ID对应亚群的信息
#这里因为我只进行了tSNE,所以这里只有一个,如果进行了tSNE和UMAP的话,这里应该是有三个,也就是PCA、tSNE和UMAP scRNA@reductions[["pca"]]@cell.embedding#细胞在PC轴上的坐标(感觉没用到过) scRNA@reductions[["tsne"]]@cell.embeddings#同理可得tSNE上每个细胞的坐标

晨曦解读
这里其实也可以看出来,PCA和tSNE都是细胞聚类,并不是涉及到表达量的变化,所以储存的大多数都是位置坐标
所以可以总结
表达数据在assays、质控信息在meta.data、分组信息在active.ident以及位置信息在reductions
Seurat的前世今生(Seurat 4.0 vs Seurat 3.0)
晨曦解读
其实概括起来4.0和3.0之间除了部分参数确实发生了变化,其主要的区别大概有四点
1. Integrative multimodal analysis即允许多形式分析,支持ncluding CITE-seq, ASAP-seq, 10X Genomics ATAC + RNA, and SHARE-seq等分析
2. 参考数据集可以映射到测试数据集上,即可以通过Seurat完成细胞注释
3. 4.0提高了分析速度和对内存的要求
4.更新的目的在于提高用户体验,3.0可以完美过度4.0,即使是3.0构建的Seurat对象也可以运用到4.0上,并不会出现报错
晨曦解读
关于4.0的一些参数修改
FindNeighbors函数:修改了这个函数的一些默认参数(annoy),因为发现这个默认参数对于提高了用户分析的内存要求,并且对结果的影响无法忽略,用户可以通过nn.method参数返回上一代默认设置
FindMarkers函数:重组了这个函数的代码使其更好理解(当然晨曦笔记都是根据4.0写的,所以不需要担心版本的问题)
CCA:合并数据集的计算进行了微小的变化(就是更快速内存消耗减少)
SCTransform:这个可以运用在多数据集联合分析中,并且运行的效果和速度都很令人满意
然后同时4.0也移除了一些功能(可以看一眼,需要的时候找对应的即可,基本上都是有些不用的就移除,好点的就给他换了一个位置)
Seurat Command List
#走常规的标准流程 pbmc.counts <- Read10X(data.dir = "~/Downloads/pbmc3k/filtered_gene_bc_matrices/hg19/") pbmc <- CreateSeuratObject(counts = pbmc.counts) pbmc <- NormalizeData(object = pbmc) pbmc <- FindVariableFeatures(object = pbmc) pbmc <- ScaleData(object = pbmc) pbmc <- RunPCA(object = pbmc) pbmc <- FindNeighbors(object = pbmc) pbmc <- FindClusters(object = pbmc) pbmc <- RunTSNE(object = pbmc) DimPlot(object = pbmc, reduction = "tsne")
晨曦解读
然后我们接下来分别使用我们的command来看看我们究竟可以对Seurat进行何种操作?
Seurat Object Interaction
#获取细胞ID(名称) colnames(x = scRNA) #base包 Cells( object= scRNA) #Seuratobject包 #两个函数的区别在于,一个属于base包,一个属于Seuratobject包,但是返回的结果是一样的
#获取基因ID(名称) rownames(x = scRNA)
#获取细胞ID和基因ID的数量 ncol(x = scRNA)(细胞ID的数量) #2638 nrow(x = scRNA) #13714
#获取细胞ID和亚群的对应关系 Idents( object= scRNA)
#获取此时亚群的因子水平 levels(x = scRNA)
#在Seurat中创建一个旧的亚群信息,并且把当前亚群放进去(只放置亚群信息并没有与之相对应的细胞ID) scRNA[[ "old.ident"]] <- Idents( object= scRNA) scRNA <- StashIdent( object= scRNA, save.name = "old.ident") #上面两个代码的结果是一样的,只不过思路不一样~

#更改所有的亚群名称 Idents( object= scRNA) <- "CD4 T cells" #仅仅更改前十个细胞的亚群名称 Idents( object= scRNA, cells = 1: 10) <- "CD3 T cells"
#设置细胞亚群名称为原始的亚群名称(这个一般比较宽泛,而且也作为我们样品的名称) Idents( object= scRNA, cells = 1: 10) <- "orig.ident"#更改前十个细胞 Idents( object= scRNA) <- "orig.ident"#更改所有细胞
晨曦解读
和上面那个结果一样,这里就不展示啦~
#按照idents获取细胞ID WhichCells(scRNA,idents = c(1,2))
#按照基因表达的量获取细胞ID(通过参数可以指定是从counts、 data还是scale. data中获取) WhichCells(scRNA,expression = MS4A1> 0,slot = "data")
#更改亚群的名称 scRNA <- RenameIdents( object= scRNA, `CD4 T cells` = "T Helper cells")
#搜索指定亚群的细胞ID数量(返回的是一个summarize) subset(x = scRNA, idents = "CD3 T cells") #这里有10个细胞ID(样本)是CD3 T cells这个亚群的,一共有13714个Gene,2000个高可变基因,2个降维聚类(PCA和tSNE) #如果我们想要真正的作用在我们的Seurat对象上,我们只需要进行赋值即可实现 #赋值要慎重,所以后面我们都是没有赋值只是单纯的看结果
#搜索指定细胞亚群,根据参数不同可以改变搜索模式 subset(x = scRNA, idents = c( "CD4 T cells", "CD3 T cells"), invert = FALSE) #输出是CD4 T cells亚群或CD3 T cells的summarize(PS,一共就2638个细胞,所以这里全部输出了出来) subset(x = scRNA, idents = c( "CD4 T cells", "CD3 T cells"), invert = TRUE) #输出既是CD4 T cells亚群也是CD3 T cells的summarize
#输出符合Gene表达的样本信息(返回的是summarize) subset(x = scRNA, subset = MS4A1 > 3) subset(x = scRNA, subset = MS4A1 > 3 & PC1 > 5) #这里的PC值得是PC指数(其实这个很少用,也不用细究) subset(x = scRNA, subset = MS4A1 > 3, idents = "B cells") #如果我们想要真正的作用在我们的Seurat对象上,我们只需要进行赋值即可实现 #赋值要慎重,所以后面我们都是没有赋值只是单纯的看结果

subset(x = scRNA, subset = orig.ident == "pbmc3k")
subset(x = scRNA, downsample = 100)
# Merge two Seurat objects merge(x = pbmc1, y = pbmc2) # Merge more than two Seurat objects merge(x = pbmc1, y = list(pbmc2, pbmc3))
晨曦解读
这里因为没有太多Seurat对象就不展示啦,其实这就是单细胞数据多数据集联合分析中的多数据集合并(区别于多数据集整合哦)
其实这里的object interaction更多的是让我们观察这个Seurat对象,从中获得更多的信息~
Data Access
#获取Seurat对象中的信息 scRNA[[]] #变量:样本信息、RNA数量、基因数量、线粒体比例、resolution参数为0.5的时候亚群标号、最终亚群编号、旧的亚群编号 #观测:细胞ID
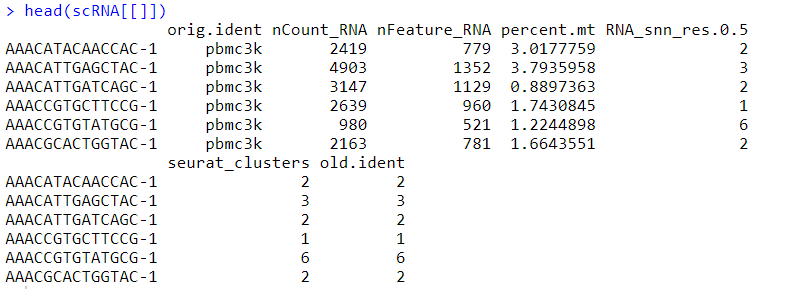
#从Seurat对象中提取特定信息 scRNA $nCount_RNA pbmc[[c( "percent.mito", "nFeature_RNA")]] #这个其实在数据集页面操作更简单

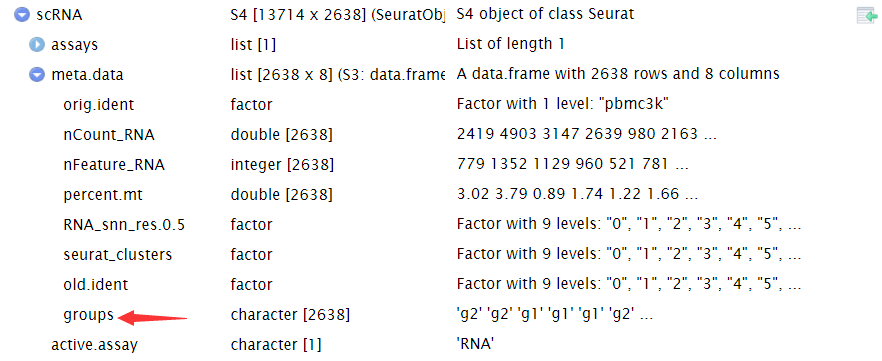
#随机添加分组信息并且把分组信息添加到Seurat对象中 random_group_labels<- sample(x = c( "g1", "g2"), size = ncol(x = scRNA), replace = TRUE) scRNA $groups<- random_group_labels
#提取Seurat对象中的原始表达矩阵(参数可以选择counts(原始)、data(标准化后)、scale.data(高可变基因)) GetAssayData( object= scRNA, slot = "counts") #对特定数据进行更新 scRNA <- SetAssayData( object= scRNA, slot = "scale.data", new.data = new.data) #这个函数就是对scRNA对象中的scale.data进行更新,这回则是数据的彻底覆盖,并不会令出现一个对象
SetAssayData函数之前
SetAssayData函数之后
#获取样本的PC信息(我们会根据这些数据来选择合适的PC,当然这个其实也不用看懂,因为我们后期会有可视化来选择合适的PC)(一般都是分50个PC) Embeddings( object= scRNA, reduction = "pca") #横坐标是细胞ID
Loadings( object= scRNA, reduction = "pca") #横坐标是Gene ID head(Loadings( object= scRNA, reduction = "pca"))[ 1: 5, 1: 6] #获取前5个Gene的前6个PC
#提取Seurat对象中的特定信息(可以选择Gene、线粒体比例、PC等等) FetchData( object= pbmc, vars = c( "PC_1", "percent.mito", "MS4A1")) #重点为返还的形式,返还的是一个类似数据框的形式,横坐标为细胞ID,纵坐标为想要的特定信息 #含义为特定信息在每个细胞ID内的情况
晨曦解读
这部分就是对Seurat对象的提取~
Visualization In Seurat
晨曦解读
这部分其实就是对Seurat对象中的某些数据进行可视化,毕竟Seurat包中也包含了很多优秀的绘图函数,当然我们也可以通过把Seurat对象中的数据提取出来用ggplot2来绘图,这也是可以的~
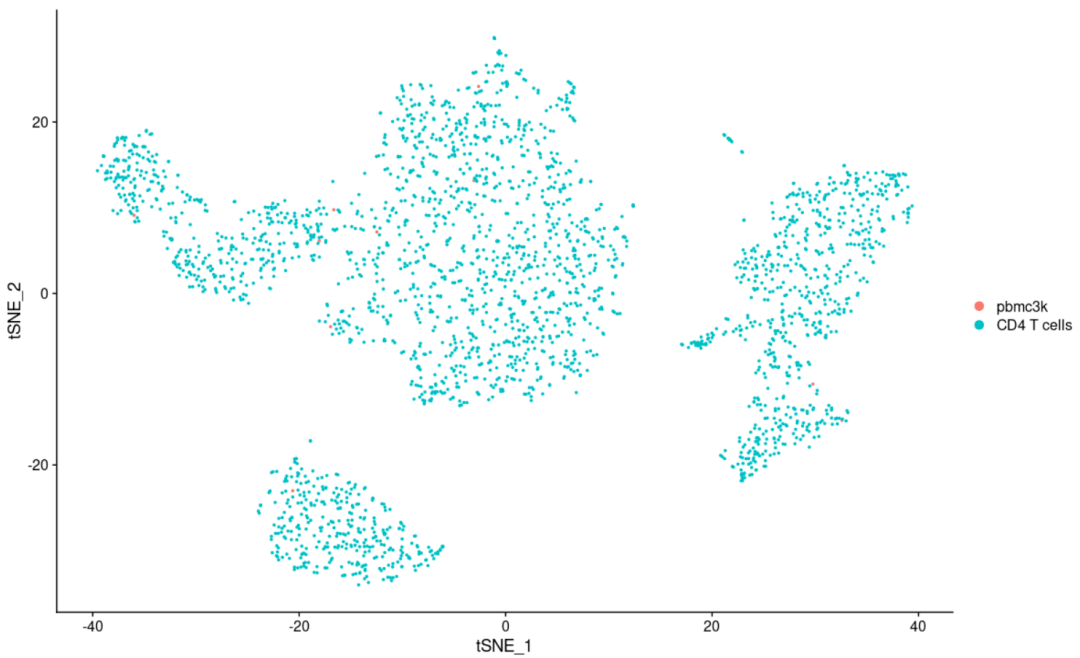
#聚类图 DimPlot( object= scRNA, reduction = "tsne") #常用 DimPlot( object= scRNA, reduction = "pca") #很少用,基本上只要知道可以绘制就可以
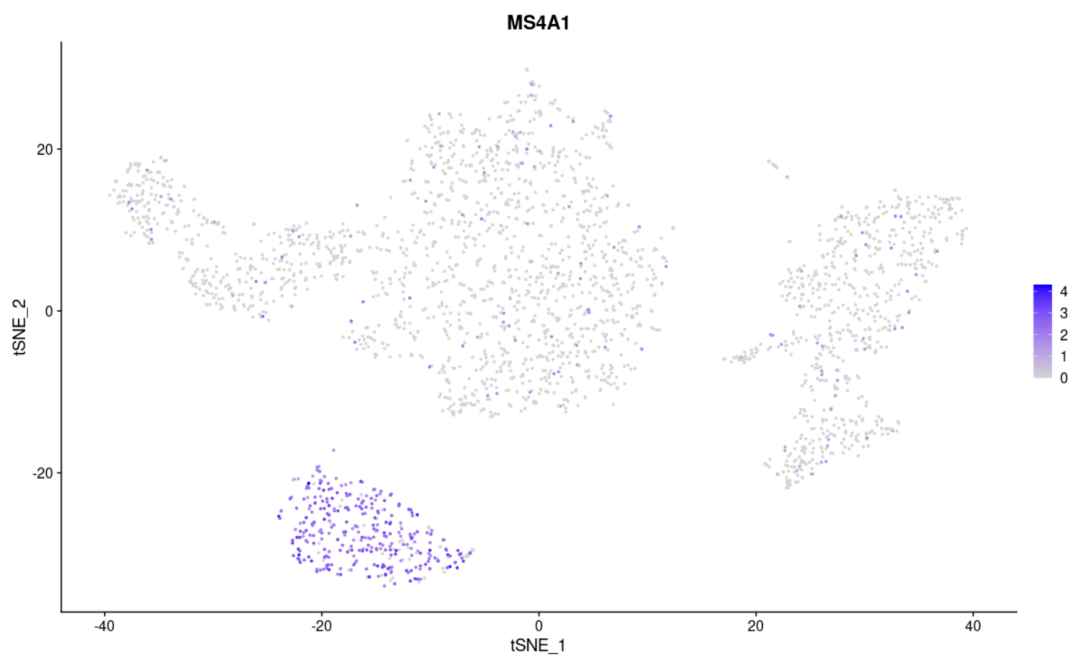
#基因在聚类图上的表达 FeaturePlot( object= scRNA, features = "MS4A1")
#两个变量相关性散点图 FeatureScatter( object= scRNA, feature1 = "MS4A1", feature2 = "CD3D")
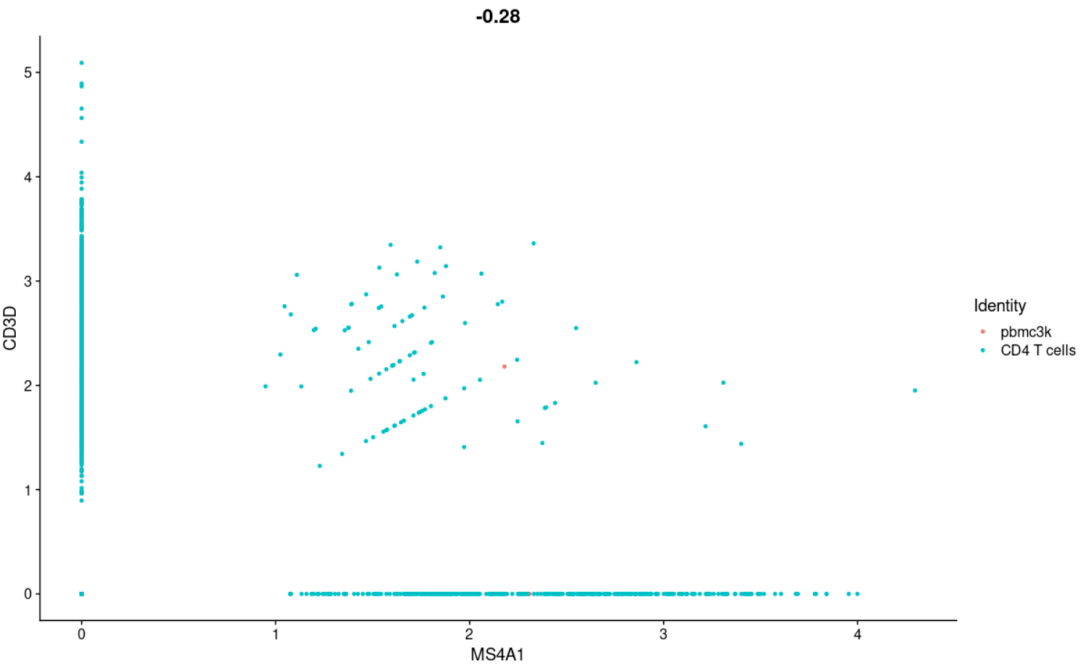
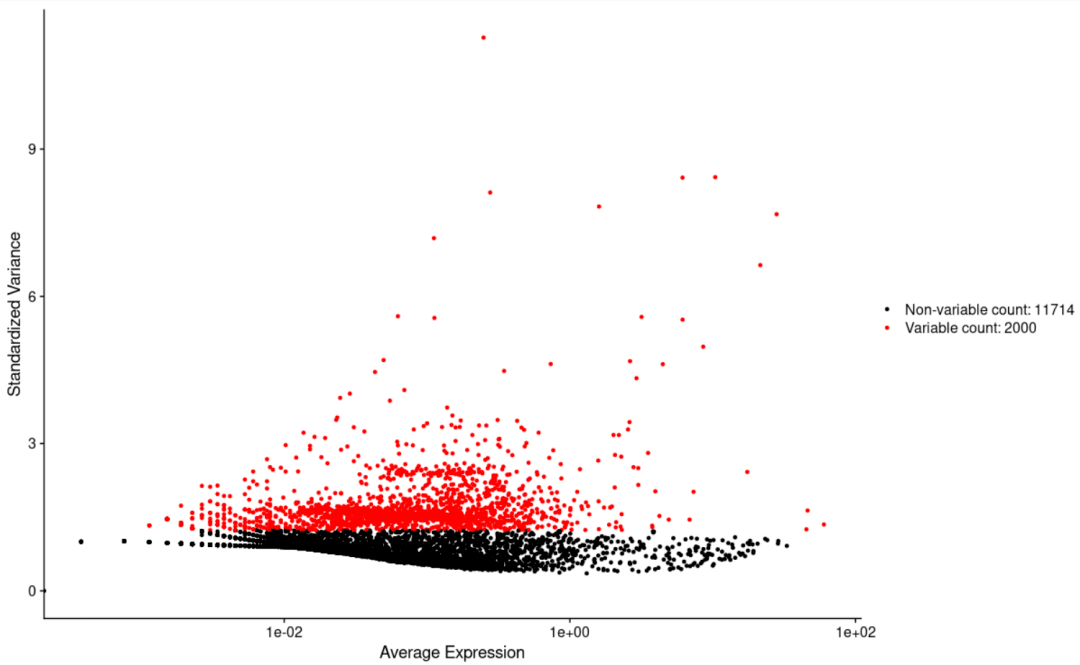
#绘制两个变量的相关性点图(这里是细胞ID) CellScatter( object= scRNA, cell1 = "AGTCTACTAGGGTG", cell2 = "CACAGATGGTTTCT") #基本不常用,估计是想评价个别样本之间的质量才会用到这个吧 #绘制高可变基因的展示图 VariableFeaturePlot( object= scRNA)
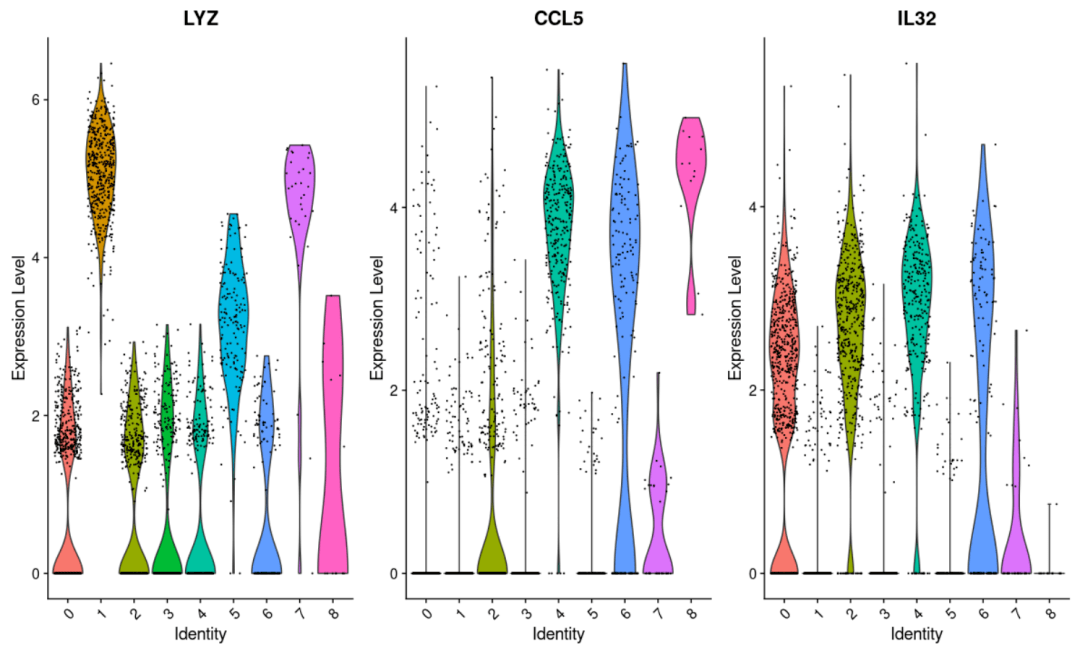
#展示基因在细胞亚群中的表达 VlnPlot( object= scRNA, features = c( "LYZ", "CCL5", "IL32")) VlnPlot( object= scRNA, features = "MS4A1", split. by= "seurat_clusters") #本质上来说结果是一样的,默认为亚群作为分组,当然你也可以自己修改,通过修改split.by参数
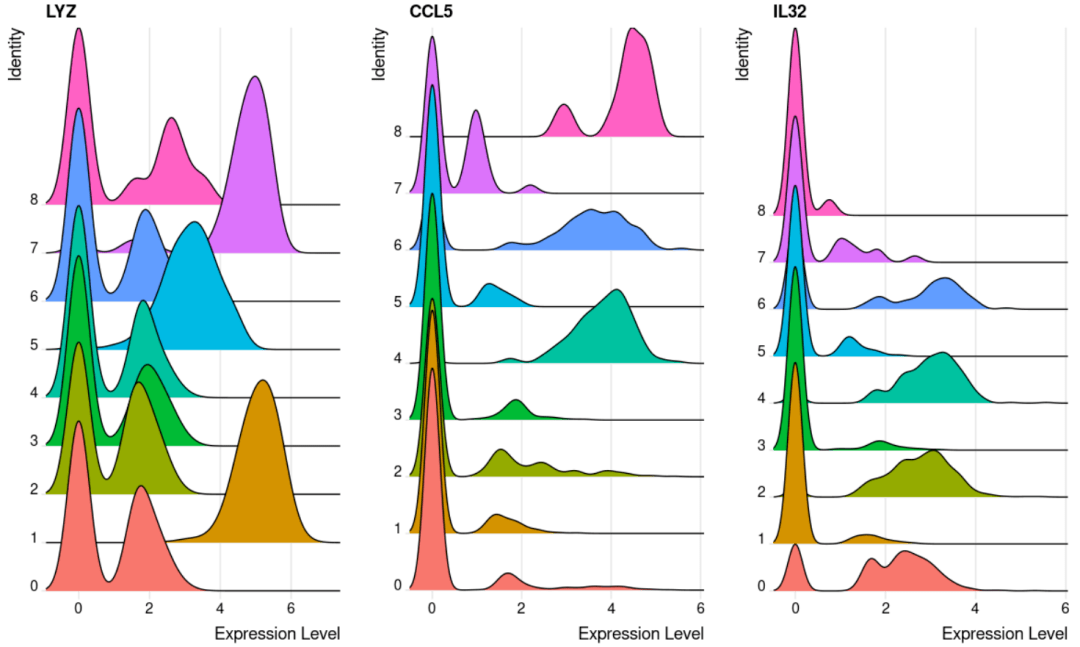
#展示基因在多个细胞亚群中的表达 RidgePlot( object= scRNA, feature = c( "LYZ", "CCL5", "IL32"))
#展示基因在多个细胞亚群中的表达 DoHeatmap( object= scRNA, features = c( "LYZ", "CCL5", "IL32"))
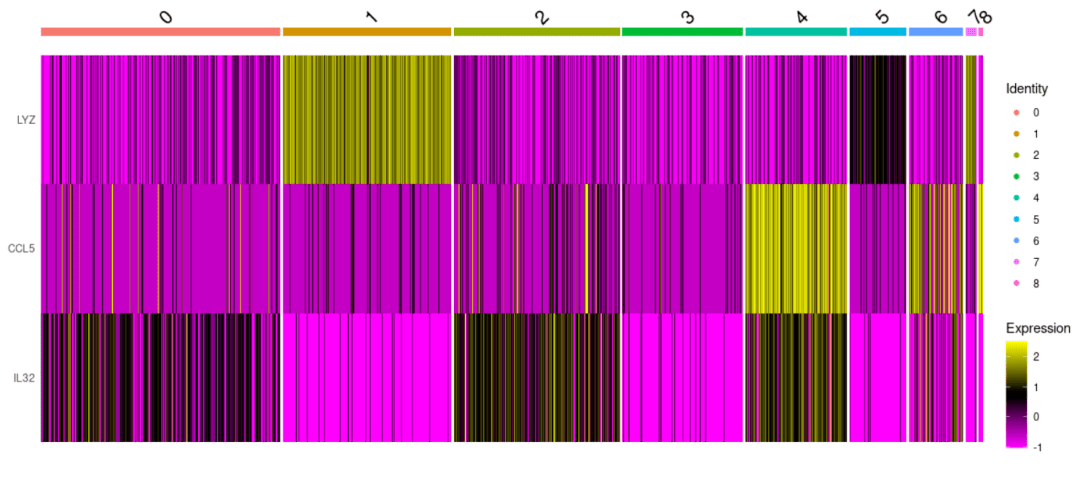
#展示当前PC下基因的表达(只要分布的鲜明,证明这个PC还是可以很好的区分) DimHeatmap( object= scRNA, reduction = "pca", cells = 200)

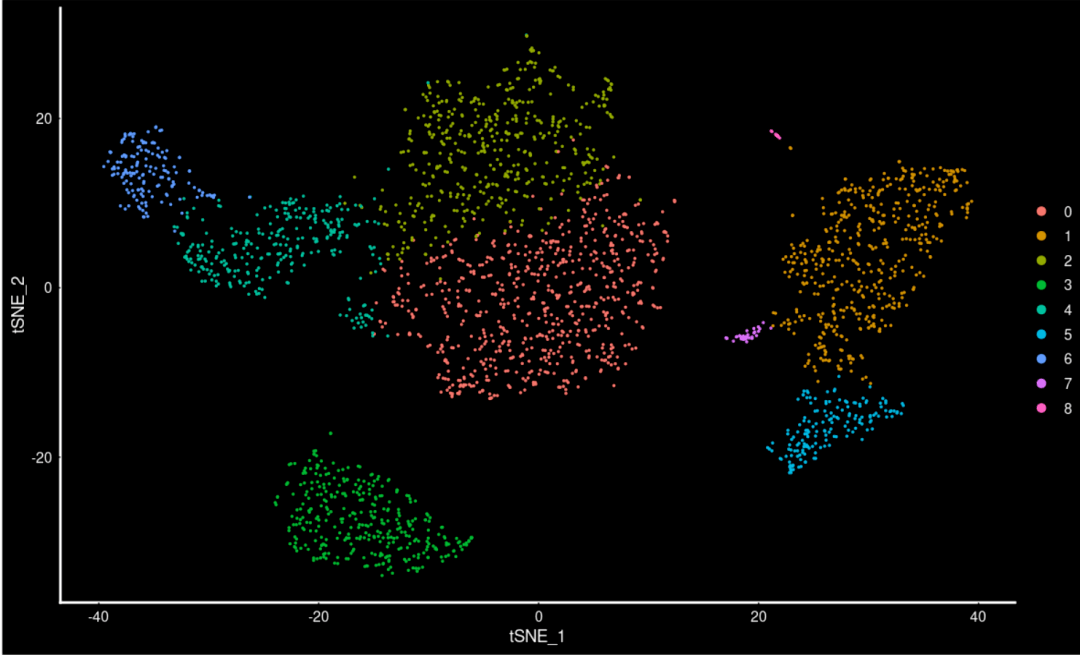
#和ggplot2联动来修改吧~ DimPlot( object= scRNA) + DarkTheme
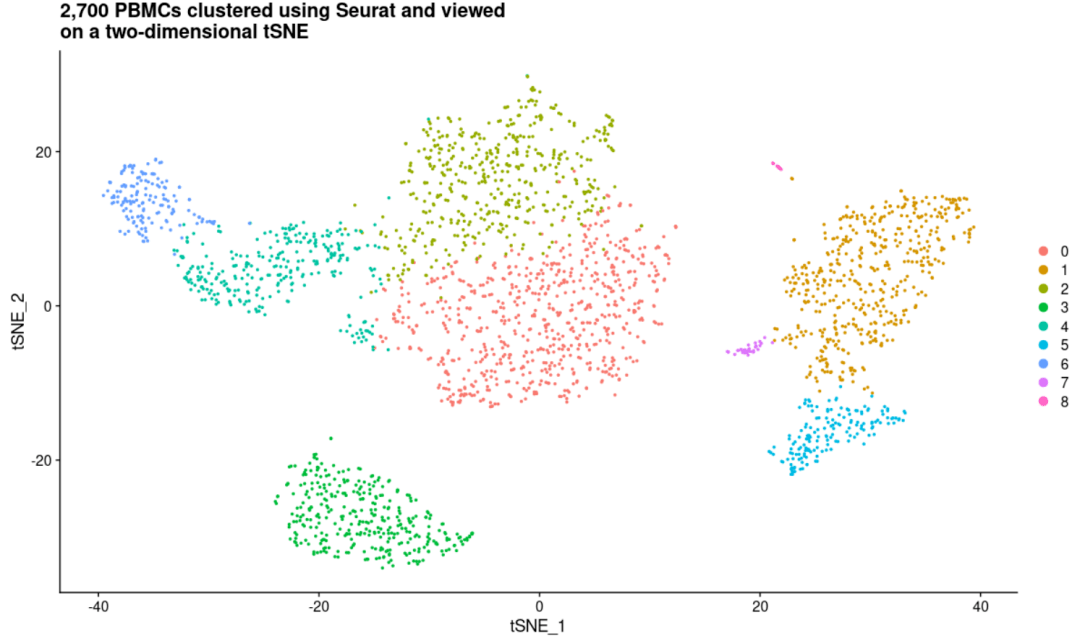
DimPlot( object= scRNA) + labs(title = "2,700 PBMCs clustered using Seurat and viewed\non a two-dimensional tSNE")
晨曦解读
Seurat提供了很多一键式的主题,可以帮助我们更好的DIY我们的可视化,同时支持了和ggplot2的联动后,为我们后续的可视化提供了方便
下面主题的使用方式为:
#举例 DimPlot( object= scRNA) + NoAxes


#去掉图例 plot <- DimPlot( object= scRNA) + NoLegend
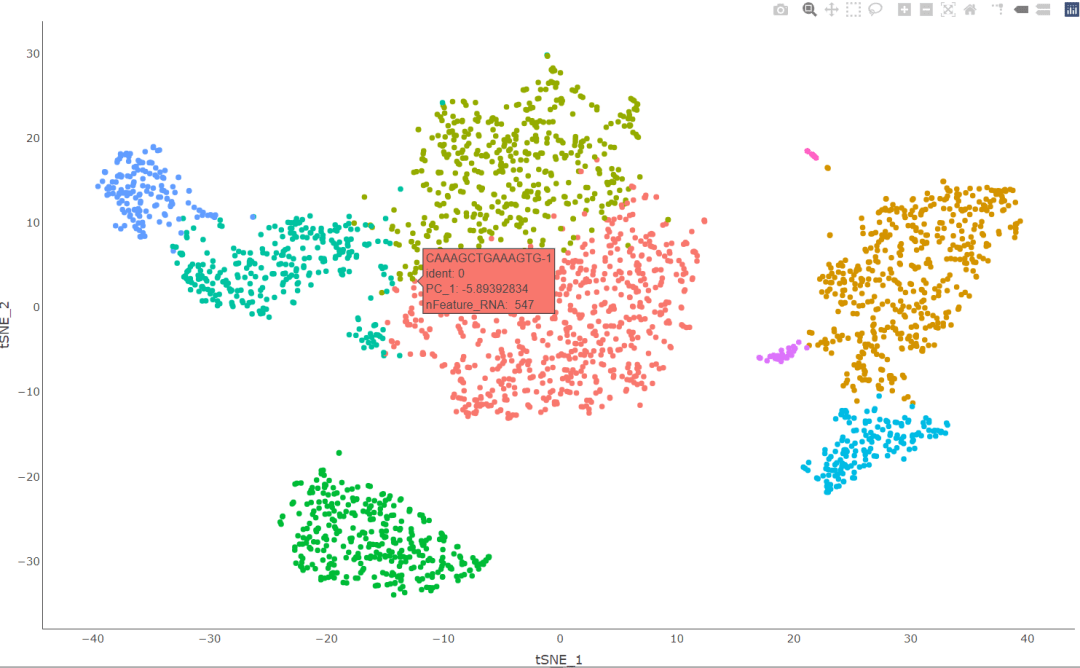
#创造交互页面 HoverLocator(plot = plot, information = FetchData( object= scRNA, vars = c( "ident", "PC_1", "nFeature_RNA"))) #其中括号内的变量会在我们移动鼠标的时候在该细胞所在的地方体现出来
#也是调出一个交互式页面,感觉没啥用处 select.cells <- CellSelector(plot = plot)
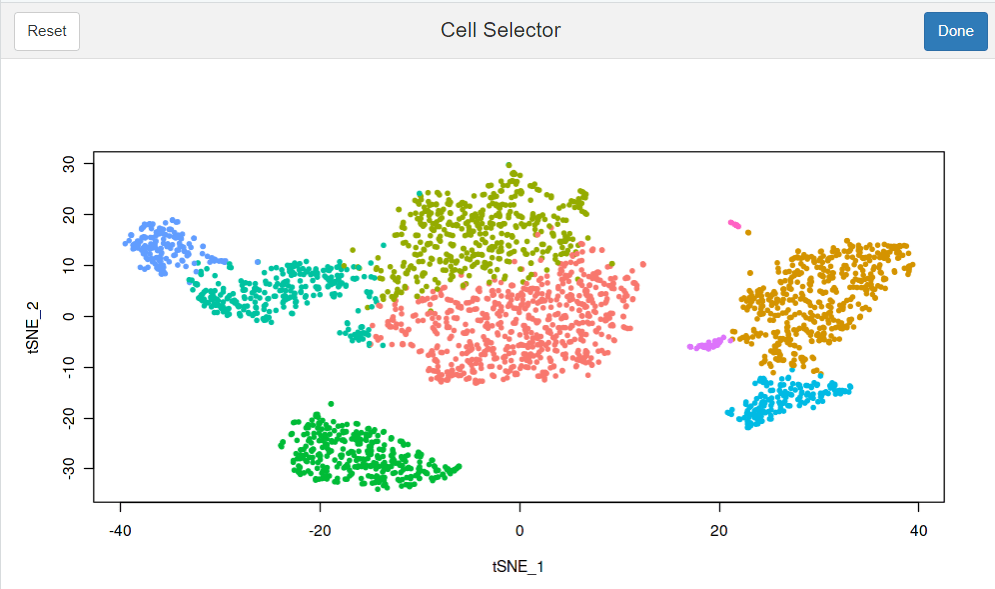
#对当前图片上的点进行标记(这个图可以是tsne,也可以是umap或者是pca,需要哪个图,把图片赋值给plot即可) LabelPoints(plot = plot, points = TopCells( object= pbmc[[ "pca"]]), repel = TRUE) #Topcells函数是返回贡献最大的基因列表,这里应该是返回贡献最大的细胞ID #repel参数是为了防止标签重叠的
晨曦解读
本来官网还有一块是Seurat现在支持多个数据模式的切换,但是因为现在没涉及到,而且最多的平台就是10×,所以暂时跳过~
Seurat对象的灵活运用
(基于前面的知识讲解,算得上是进阶版吧)
晨曦解读
首先我们载入走过标准流程的RDS文件,然后为了模拟有两个重复的情况,我们把其分开,一个命名为“rep1”,一个命名为“rep2”
#准备工作 library(Seurat) scRNA <- readRDS( "~/scRNA.rds") #分成两组 set.seed( 42) scRNA$replicate <- sample(c( "rep1", "rep2"), size = ncol(scRNA), replace = TRUE) #这个其实就是在meta.data里添加了一个变量replicate,然后作为我们后续的分组信息

#从细胞聚类和样本分组中切换idents(其实就是把样本分组映射到细胞聚类上来) DimPlot(scRNA, reduction = "tsne")
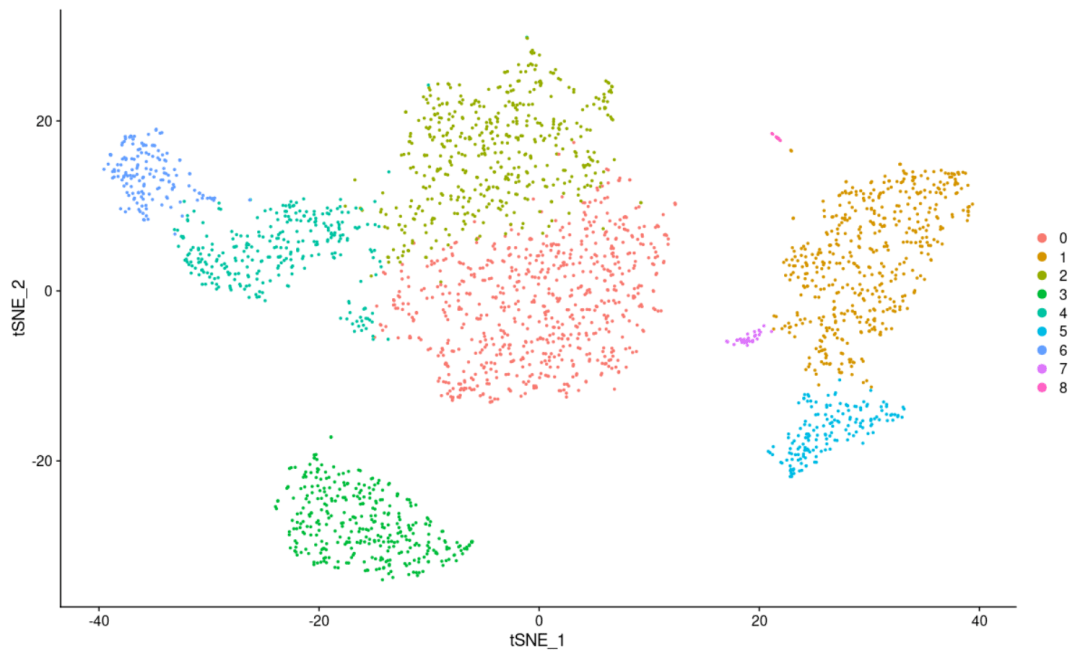
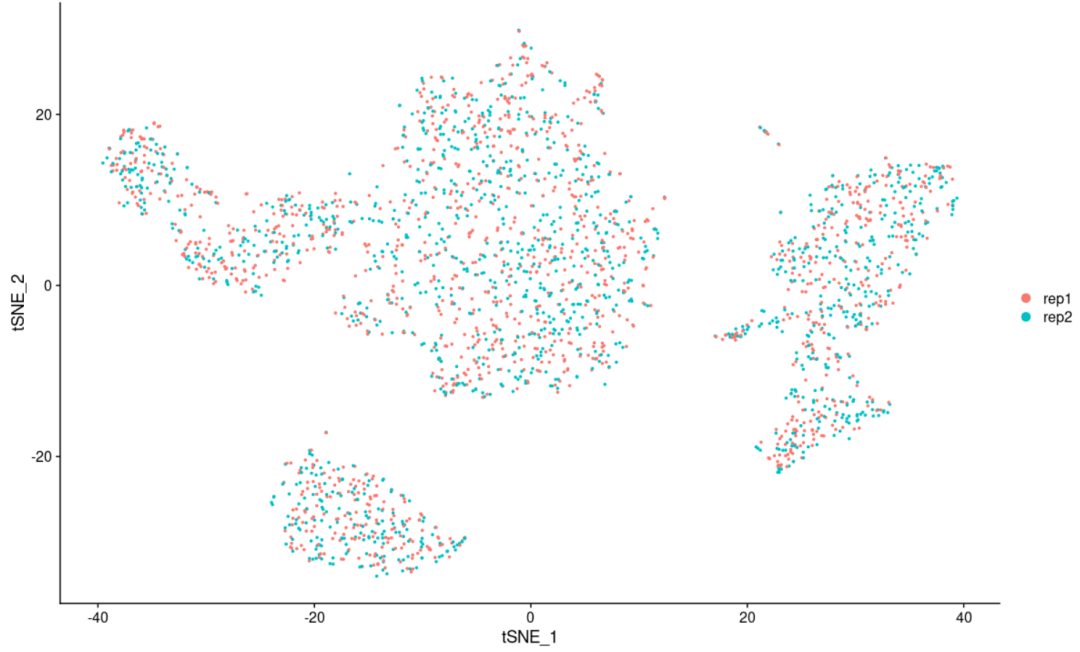
#首先把我们目前的细胞亚群分类储存到对象中 scRNA$CellType <- Idents(scRNA) #接下来调取我们的分组信息到对象中 Idents(scRNA) <- "replicate" DimPlot(scRNA, reduction = "tsne") #和下面的代码执行的是一件事情,因为我们提前设置了亚群分组信息,所以这里我们可以直接调用 DimPlot(scRNA, reduction = 'tsne', group. by= 'replicate') #这几步的目的其实就是把我们的细胞聚类图以样品分组的形式展示,可以展示的还有很多,比如各个样品的组织来源或者是疾病状态,反正只要是分组就可以 #绘图完毕后,我们要切换会细胞亚群的形式(可以简单理解为Seurat对象中储存着很多分组信息,默认是细胞聚类信息,但是我们通过设置可以设置成其它的分组信息,并且可以映射到细胞聚类的可视化上面) Idents(scRNA) <- "CellType"
#上一个小节,是告诉我们如何把样本分组信息映射到细胞聚类的可视化上面 #下面这个小节,告诉我们如何同时统计不同聚类或者不同样本来源的细胞数目? #首先每个聚类包含多少细胞? table(Idents(scRNA))
#每个样本分组有多少个细胞呢?(两种方法,借此机会可以体会到上面那个“开关”效应) #方法一 table(scRNA $replicate) #方法二 Idents(scRNA) <- "replicate" table(Idents(scRNA))
#每个聚类细胞数占比(就是这个亚群的细胞占总细胞数量的比例) prop.table(table(Idents(scRNA)))
#让我们把样本分组和细胞聚类放在一起统计吧 table(Idents(scRNA), scRNA $replicate)

prop.table(table(Idents(scRNA), scRNA $replicate), margin = 2)
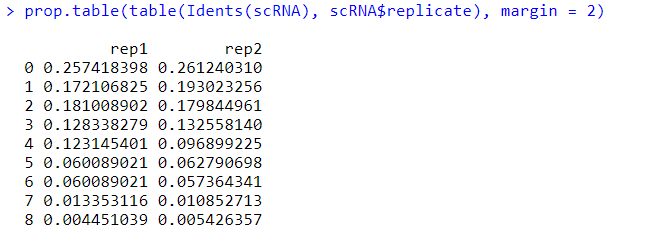
#第一个小节:样本分组信息映射到细胞聚类 #第二个小节:展示各种细胞数目和百分比 #第三个小节:我们将开始学习提取特定的Seurat子集来供我们进行后续的分析 #这里本来要选择细胞亚群的名字比较好,但是我选择的这个实例数据集还没有进行亚群注释,所以暂时用编号代替,其实原理是一样的 WhichCells(scRNA, idents = "0") #会显示出该细胞亚群(注释)下所属的细胞ID
#接下来我们想要提取出该细胞亚群所属细胞的原始表达矩阵 raw.data <- as.matrix(GetAssayData(scRNA, slot = "counts")[, WhichCells(scRNA, ident = 0)])
#然后我们要获取某基因表达量大于1的样本数 subset(scRNA, subset = MS4A1 > 1)
#想要了解属于某个分组的细胞数量 subset(scRNA, subset = replicate == "rep2")
#探索:我们是否可以只创建一个含有某种细胞类型的对象呢? subset(scRNA, idents = c( "0", "1"))
#探索:我们是否可以创建一个不包含某类细胞亚群的对象呢? subset(scRNA, idents = c( "0", "1"), invert = TRUE)
#第四节:我们来探索如何计算基因的平均表达量(即一个基因在众多细胞 ID中的平均表达量) cluster.averages< -AverageExpression( scRNA) #返回的是一个列表,里面只有一个数据,然后我们把这个数据提取一下发现是某个基因在某个细胞亚群中的平均表达量 cluster.averages[["RNA"]] head( cluster.averages[["RNA"]] [, 1:5])
#开启下游分析(可视化) #绘图的时候空格容易失败,所以我们接下来需要把我们分组信息(亚群注释)中带空格的给替换成 "_" orig.levels <- levels(scRNA) Idents(scRNA) <- gsub(pattern = " ", replacement = "_", x = Idents(scRNA)) orig.levels <- gsub(pattern = " ", replacement = "_", x = orig.levels) levels(scRNA) <- orig.levels #说实话没搞懂这个弯弯绕存在的目的,如果直接下面的代码不也可以么? Idents(scRNA) <- gsub(pattern = " ", replacement = "_", x = Idents(scRNA)) #然后我们把我们的平均表达量放回进Seurat对象中(后续绘图我们就可以直接调用每个基因在每个亚群的平均表达量了) cluster.averages <- AverageExpression(scRNA, return.seurat = TRUE) #可视化(想要知道某两个细胞亚群的表达情况) CellScatter(cluster.averages, cell1 = "0", cell2 = "1") #这里没法做的和官网那么漂亮,估计是我的数据的问题,所以这里就不放图了
晨曦解读
上面这些都是挺实用的计算方式,学起来,记不住可以当成笔记,记录保存好~
#计算不同样本分组的基因平均表达(区别于上面,上面是计算基因在不同细胞亚群的表达) cluster.averages <- AverageExpression(scRNA, return.seurat = TRUE, add.ident = "replicate") #然后同样可以画出一个不同分组的基因表达相关性点图 CellScatter(cluster.averages, cell1 = "CD8_T_rep1", cell2 = "CD8_T_rep2") #这里我们也可以使用热图来承接我们的平均表达量数据 DoHeatmap(cluster.averages, features = unlist(TopFeatures(pbmc[[ "pca"]], balanced = TRUE)), size = 3, draw.lines = FALSE)
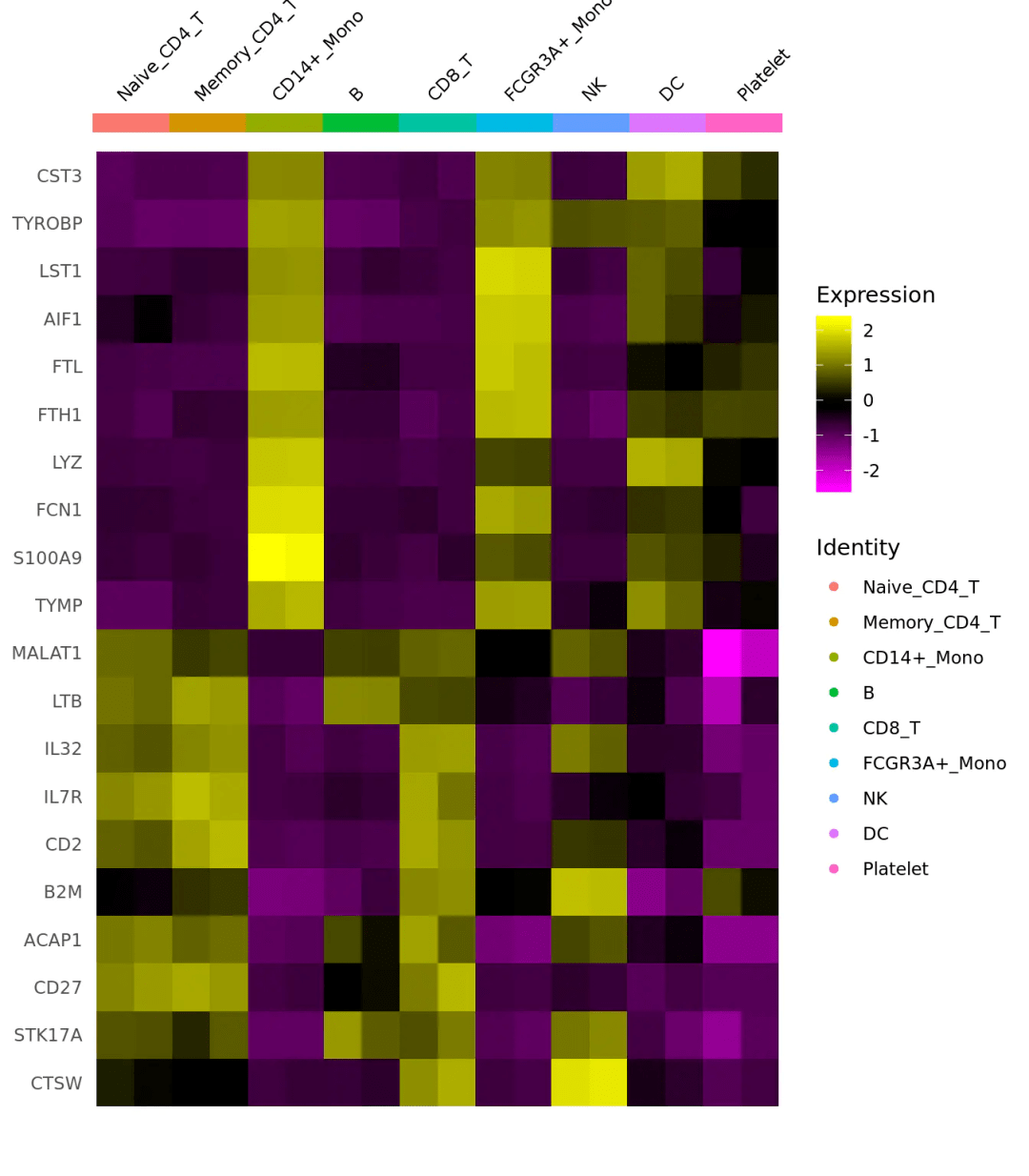
晨曦解读
接下来做一个串讲,虽然Seurat内置了很多绘图函数,可以帮助我们更好的进行可视化,甚至于,可以部分和ggplot2进行联动,但是如果我们把数据提取出来,直接用ggplot2来进行绘图岂不是更加完美,那么我们现在就来开始一下这个流程
expr <- data.frame(FetchData( object= scRNA, vars = "LYZ")) #这里我们随便选择了一个Gene,当然我们也可以选择很多变量,比如亚群、线粒体比例、PC、tSNE等等,基本上只要在Seurat对象中都可以通过这个函数给提取出来,提取出来的数据形式上面都介绍过~
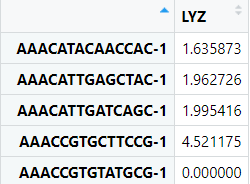
#一般来说我们绘制图形,除了表达量以外还需要一个就是分组信息,分组信息我们可以选择细胞亚群,当然也可以选择样本分组 #scRNA$replicate <- sample(c( "rep1", "rep2"), size = ncol(scRNA), replace = TRUE) #当然这里我们还是选择现成的细胞亚群 expr$Barcod<-rownames(exprs) ident<- data.frame ident<- data.frame(Barcod=names(Idents( object= scRNA)),orig.ident=Idents( object= scRNA)) c<-merge(expr,ident, by= 'Barcod')
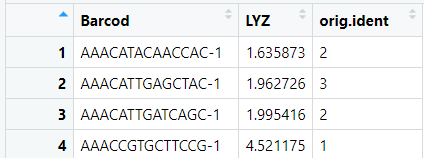
#然后开始绘图,今天就简简单单绘制一个箱线图吧 library(ggplot2) ggplot(c,aes(orig.ident,LYZ,fill=orig.ident)) + geom_boxplot
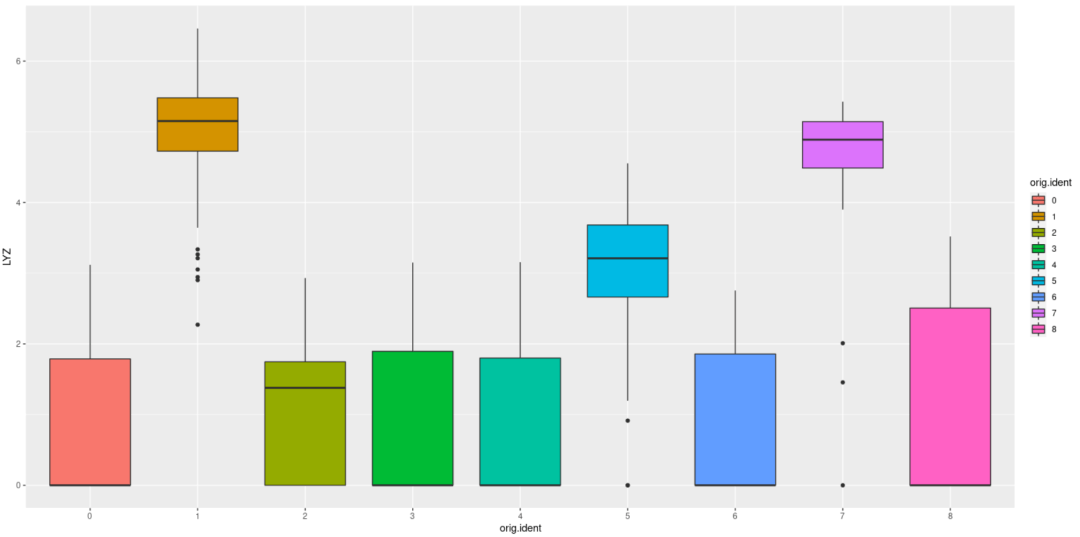
#对可视化进行修改 ggplot(c,aes(orig.ident,LYZ,fill=orig.ident)) + geom_boxplot(notch = F,width= 0.5,alpha= 0.8) + stat_summary( fun="mean",geom="point",shape=23, size= 4,fill= "white")+ stat_boxplot(geom = "errorbar",width= 0.1)+ theme_bw + coord_flip
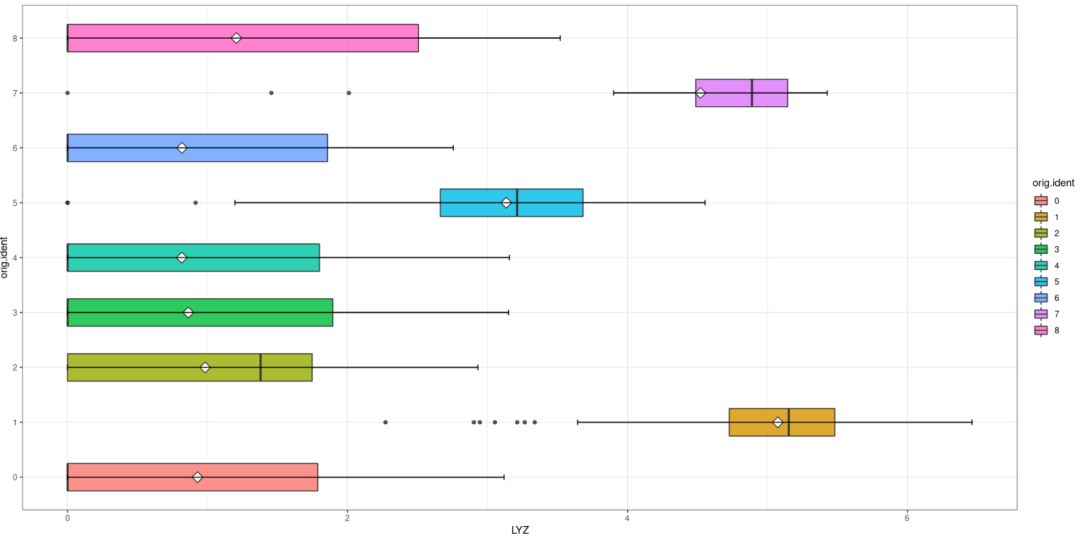
晨曦解读
把数据整理成ggplot2可以接受的形式后,单细胞数据配上功能强大的ggplot2,只能说可以碰撞出无限可能~
那么本期推文就结束啦~
经过这一篇推文,大家对Seurat的了解就足以完成后续的一些常规分析了,其实大家也可以把这篇推文当成一本书,如果遇到想要实现的命令,来这里查询,大概率都会得到解决~
下一次介绍进行一下多数据集联合方法的对比,然后再介绍几个细胞注释的方法后,我们就要开始单细胞的高级分析了,前段时间的基础越牢固,后期才会更加顺利哦
我是晨曦,我们下次再见QAQ
回复“晨曦06”,即可获得今天的范例数据和代码哦~
— END—
撰文丨晨 曦
排版丨四金兄
主编丨小雪球
转自解螺旋生信频道-挑圈联靠公号~
