近年的研究热点集中于环境和生物体相互作用的微生物群体,而大量复杂的微生物群体存在培养困难,构成复杂(包括细菌、古菌、真菌、原生生物、病毒甚至小型真核生物)。因此如何用高通量精准的了解这些群体的构成,基因功能分布以及具体的表达活性和代谢状况成为首要问题。
高通量测序技术的发展,让我们可以不经过培养,一次性了解微生物群落构成甚至基因代谢组成。
随着技术的进步,检测方法也逐渐丰富,对应的分析手段和软件算法也逐步完善,使我们可以根据研究需要选择不同的检测和分析策略来获得海量的数据并进行相应的研究分析。

01
简 介
免于培养的微生物学研究方法主要基于测序,高通量测序使我们一次可以获得整个微生物群体的数据信息,简单来说包括两种策略:
1、基于特定标记基因的扩增测序方案(常见的16s,ITs,18s或特定功能基因)
2、对整个群落DNA进行测序,获取全部微生物基因组进而进行分类和功能分析的策略(鸟枪法宏基因组测序shotgun metagenomics)。
基于16s基因的分析方法
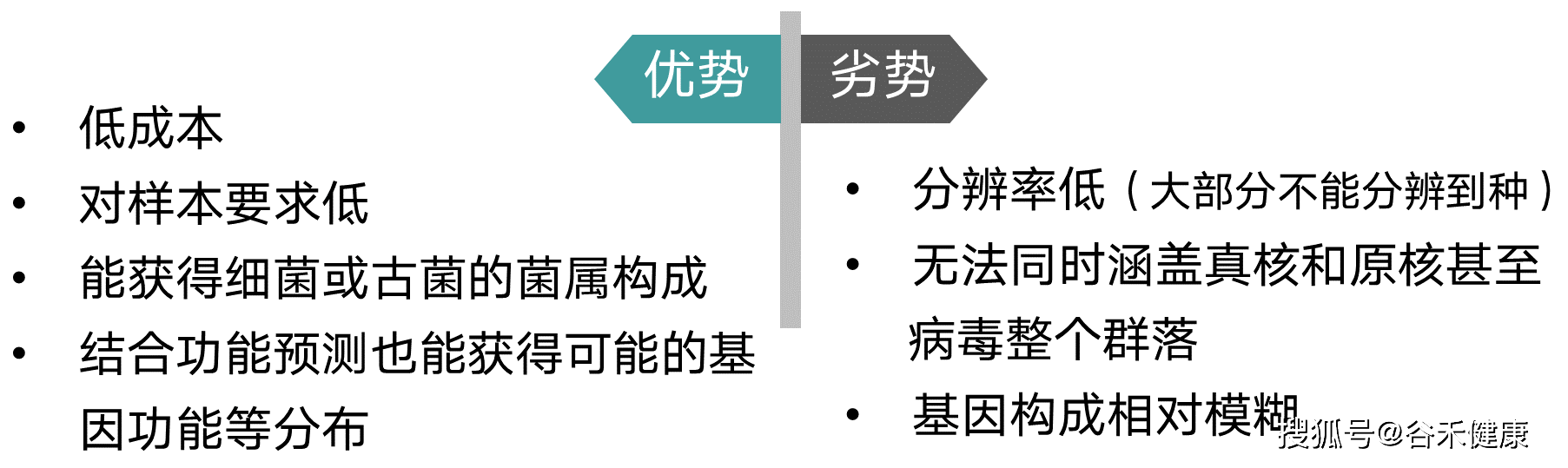
由于其极低的成本,对于样本DNA的低要求非常适合于大规模群体样本的调查和分析,随着DADA2等分析方法的改进,物种分类精度和准确度也有所提升,加上PICRUST等功能预测方法一定程度上弥补了基因信息的缺失,因此16s这类基于基因的微生物研究方法仍然是不可或缺的方案。
下表列了16s常见的分析软件,目前QIIME2作为整合包使用最为方便,VSEARCH也作为UPARSE的开源版本使用也非常广泛。
16s测序的分析流程如下图,获得序列经过聚类后获得OTU或ASV,并得到相对丰度。
经过PICRUSt可以得到预测的基因分类丰度,进而进行alpha多样性和Beta多样性以及组间差异和相关性分析。
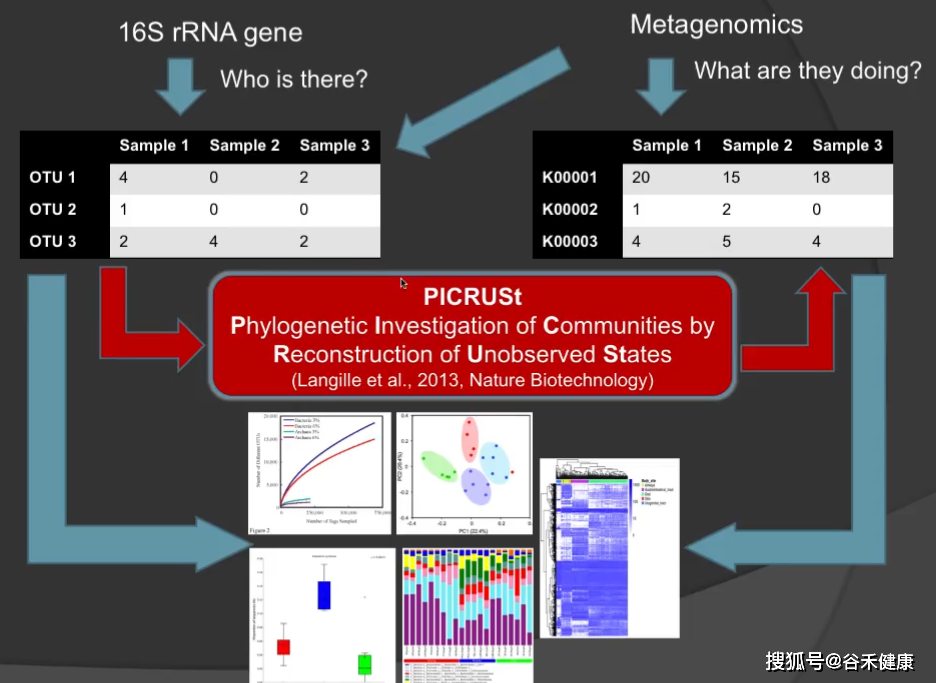
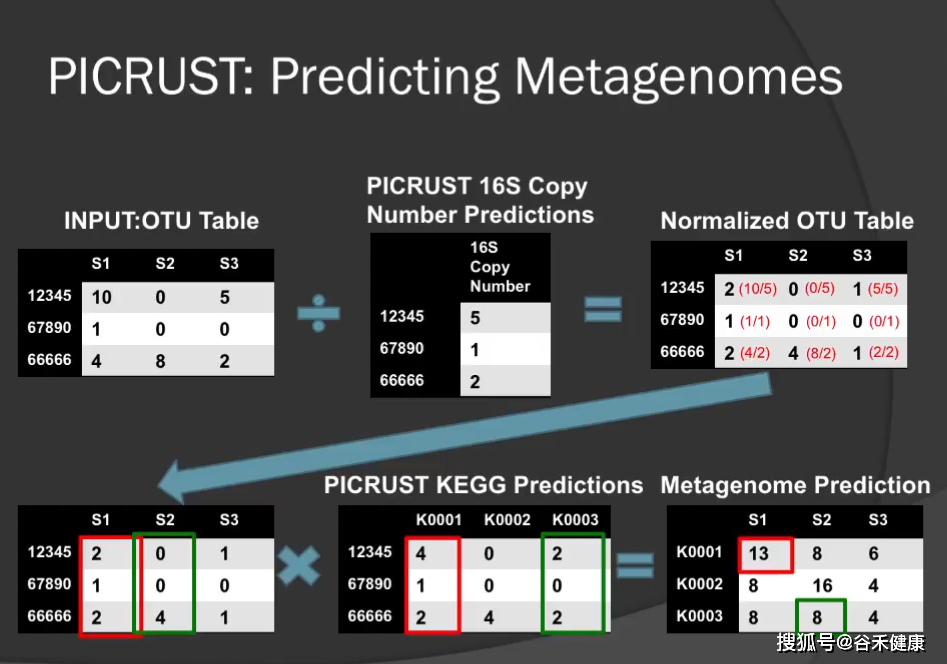
PICRSt的工作原理如下图,将OTU表内16s序列进行对应物种16s拷贝数标准化后,将物种丰度乘以已经整理好的物种的基因注释数表就获得基因的预测丰度。
02
浅宏基因组
浅宏基因组测序方案是去年knights-lab在msystems上发表的针对16s分辨率和宏基因组高成本之间的一个折中方案,通过降低测序深度,每个样本50万reads,但是物种的分辨率并没有低于一般宏基因组(普遍5~10G数据量)。
不通过拼接组装,直接基于kraken2等kmer,或MetaPhlAn2等标记基因的参考基因组方法进行种属丰度分类。结合其到菌株的物种分类和丰度数据可较16s方案下的PICRUST更加准确的预测基因构成。
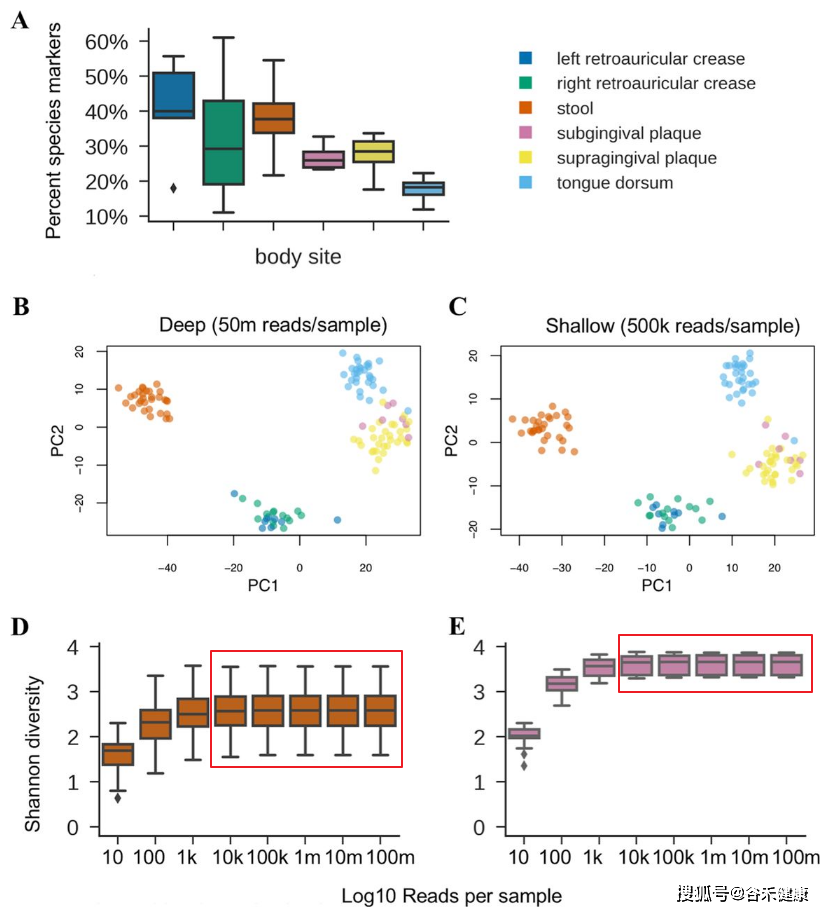
Hillmann B, Al-Ghalith GA, Shields-Cutler RR, Zhu Q, Gohl DM, Beckman KB, Knight R, Knights D. 2018. Evaluating the information content of shallow shotgun metagenomics. mSystems 3:e00069-18. https://doi.org/10.1128/mSystems.00069-18.
我们发现有些小伙伴的需求是:
想要获得更全和更精细分类精度同时不需要获得完整基因组序列和重建菌群基因的。
那么这时候,我们提供的浅宏基因组测序就可以成为很好的选择,其成本低(快要接近16s测序分析的价格了,文末有福利),分析简便快速,同样能获得宏基因组的基本丰度数据。不过浅宏基因组也有其适用范围,根据样品类型的不同,一些样品可能包含 >99%的人类宿主DNA,这不仅增加了序列成本,而且给测量带来了不确定性。
在许多研究中也会采取在进行宏基因组测序文库的准备之前去除宿主DNA的方法。但是,在去除宿主DNA后,可能没有足够的微生物基因组DNA用于宏基因组测序,这通常需要最少50ng的输入。因此浅宏基因组较适合于宿主DNA含量较低的样本,如人类粪便、水体、土壤等;而如口腔唾液、肺泡灌洗液、血液等人体体液类样本就不太适合。
下图是宏基因组测序数据中比对到人类基因组的序列比例,根据样本类型不同而不同。
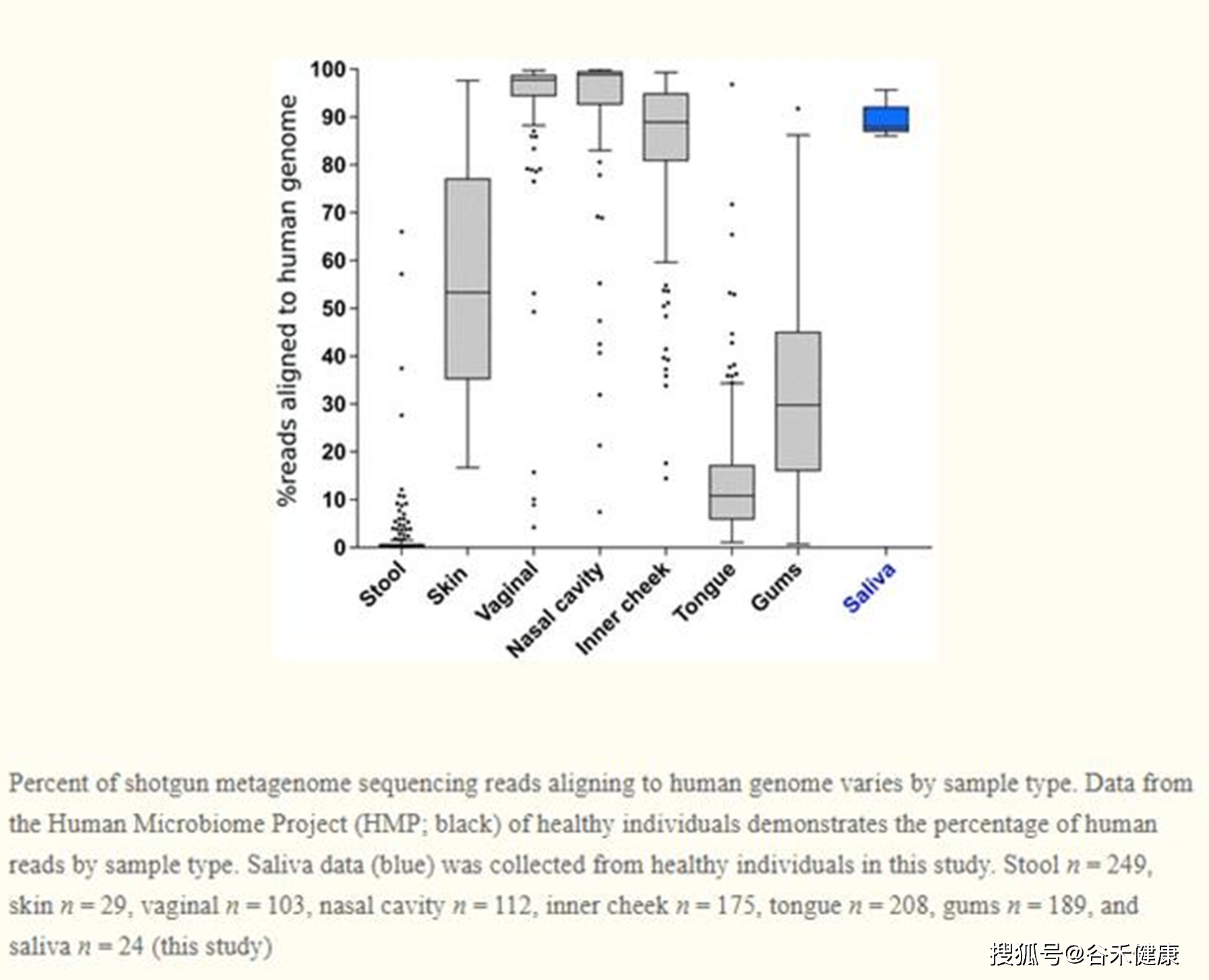
我们可以免费提供针对粪便及环境样本助力临床/科研取样。
人体口腔、痰液、腹水、脑脊液、尿液、皮肤、阴道分泌物等高寄主细胞含量样本可根据我们的处理方案简单处理后大幅降低宿主DNA比例。
处理方案如下:
- 高宿主含量DNA样本(包括唾液、血液、肺泡灌洗液、腹水、阴道分泌物和黏膜类样品)的取样前处
- 将200微升唾液等体液样本以10,000g离心8分钟
- 弃去上清液,通过移液将细胞沉淀重悬于200μl无菌水中,短暂涡旋,然后在室温下静置5分钟,以渗透压裂解哺乳动物细胞
- 添加终浓度为10μm的PMA(叠氮溴化丙锭)(向200μl样品中添加10μl的0.2 mM PMA溶液),并将样品短暂涡旋,然后在黑暗中于室温温育5分钟
- 然后将样品从标准台式荧光灯放置在<20cm的冰上水平放置25分钟,短暂离心并每5分钟旋转一次
- 完成后,可将样品冷冻在−20°C或转移到取样管的储存液中
Marotz CA, Sanders JG, Zuniga C, Zaramela LS, Knight R, Zengler K. Improving saliva shotgun metagenomics by chemical host DNA depletion. Microbiome. 2018;6(1):42. Published 2018 Feb 27. doi:10.1186/s40168-018-0426-3
本处理方案以后宿主DNA可以降低8%以下。
03
宏基因组
说起宏基因组,对于熟悉宏基因组或者打算做宏基因组的同学可能已经迫不及待想知道这个怎么分析啊,怎么看结果啊之类的问题... 但在这之前,首先你应该了解的是宏基因组是什么,做宏基因组你能得到什么。

此外,对于缺乏深度研究和高质量参考基因组的样本,如土壤和特殊环境下的样本,宏基因组获得的较为完整的基因组不仅可以丰富参考基因组数据库,同时可以提供更加准确的物种分类。
因此,深度宏基因组测序是解析新环境样本的核心方法,不过从单一样本中重建出完整的菌株基因组有相当困难,一般需要较多样本或设置梯度样本从而利用更高深度和共同变化来获取分箱信息,当然对应测序和分析成本会更高。
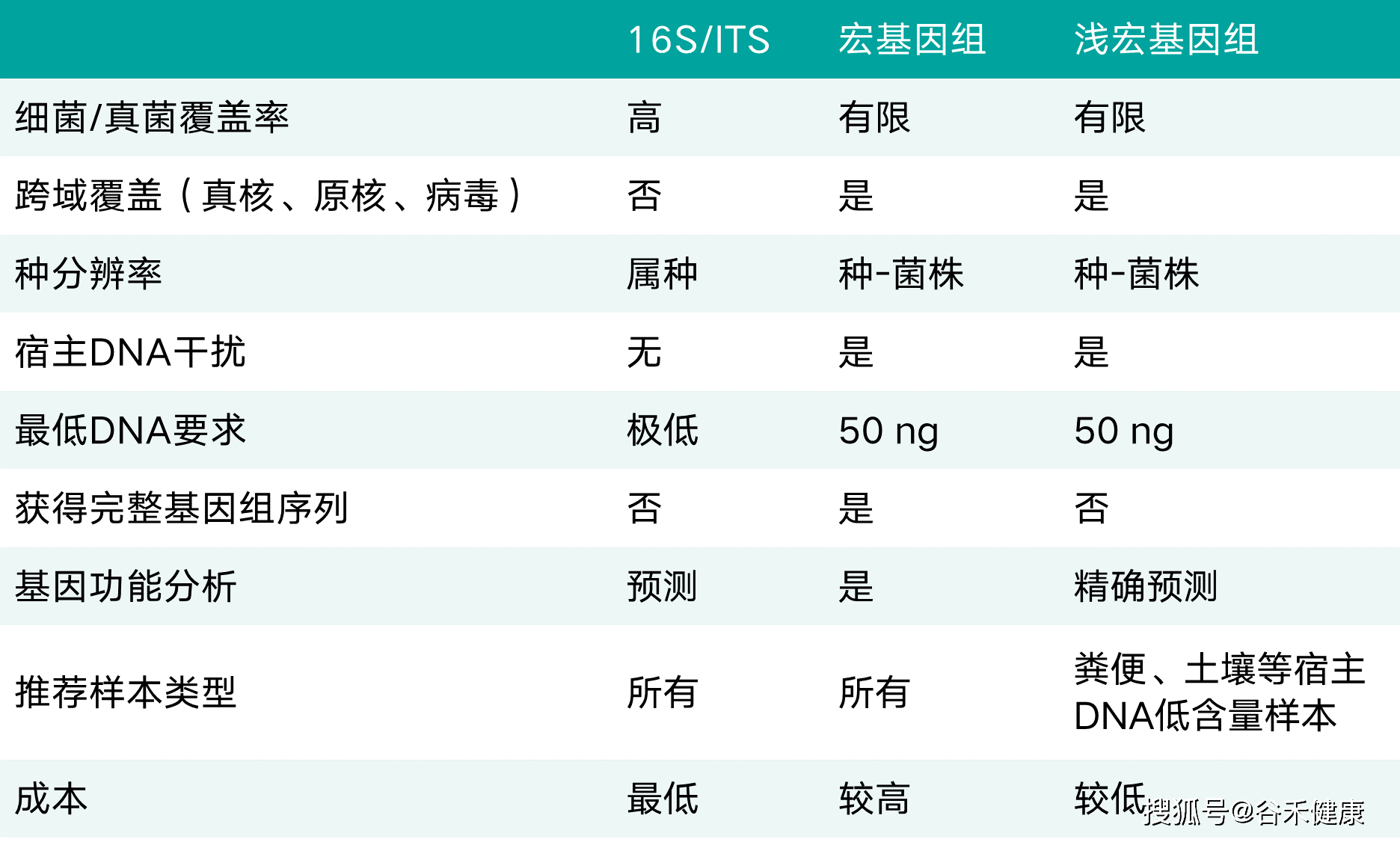
至此,我们了解了16s、浅宏基因组、宏基因组三种方式,我们将它们各自的特点总结如下表,便于你更直观地去了解(文末有福利~)。
宏基因组报告中有哪些分析内容?
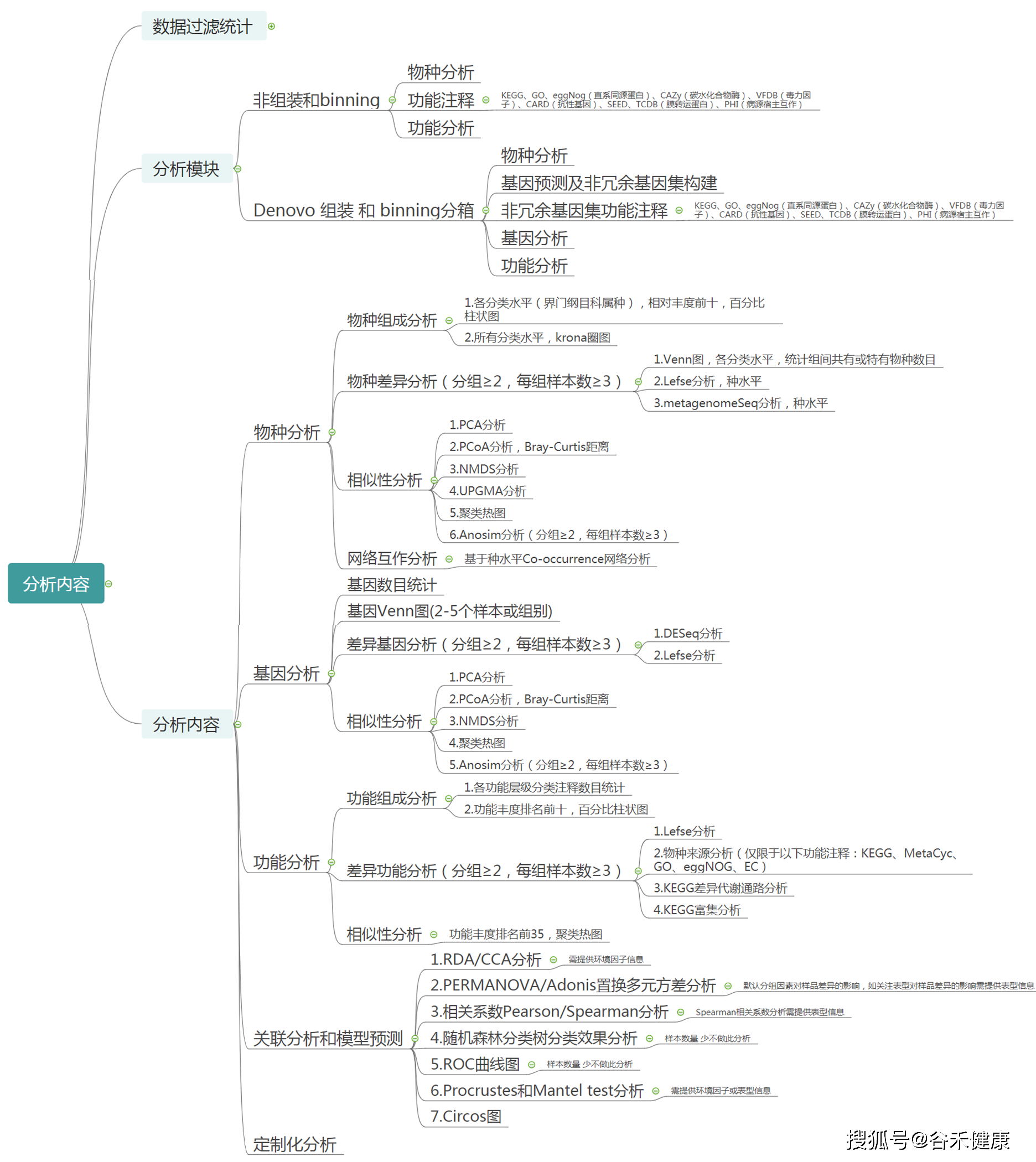
上图可以快速预览一下我们报告中的分析内容。
接下来,我们会详细介绍这些内容是如何从原始数据开始一步步实现的,同时也会选取一些文章案例来给大家做详细解读,希望给大家带来一些思路。
数据分析流程
测序数据需要经过质检,去除接头和低质量序列,一般还会进行一步过滤人的基因组序列,然后分为两个路径,使用参考数据的比对方法和从头组装的方法,下图是一个完整的宏基因组分析流程:
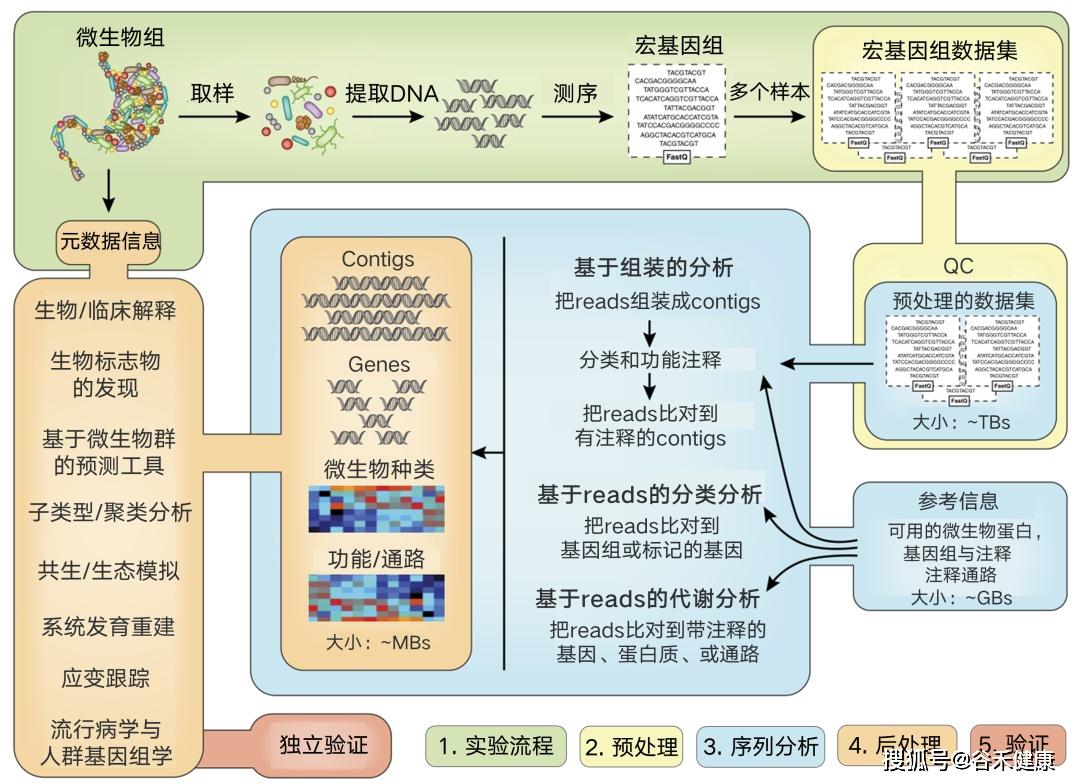
看完上图,可以对宏基因组测序的基本流程有个大致了解。
对于宏基因组测序而言,最重要的就是获得微生物群准确的物种构成及其丰度。
一、 物种构成
首先你需要了解的是无论16S测序还是宏基因组测序获得的均是相对丰度,即每种菌占所有菌属的比例。
要获得绝对的丰度需要在取样时做好取样量的计量,并在提取和建库中加入已知绝对量的参照DNA。
宏基因组测序获得物种构成及其丰度有以下两条路可以走:

我们先讲其中之一: 直接比对 。
直接比对是基于参考数据的,那么基于参考数据的物种构成分析主要有两类方法:
一类是基于Kmer和LCA比对特征来分析对应物种丰度,如kraken2等。
另一类是基于特征标记基因进行分析的,如MetaPhlAn2等。
基于参考基因组的分析工具如下表:
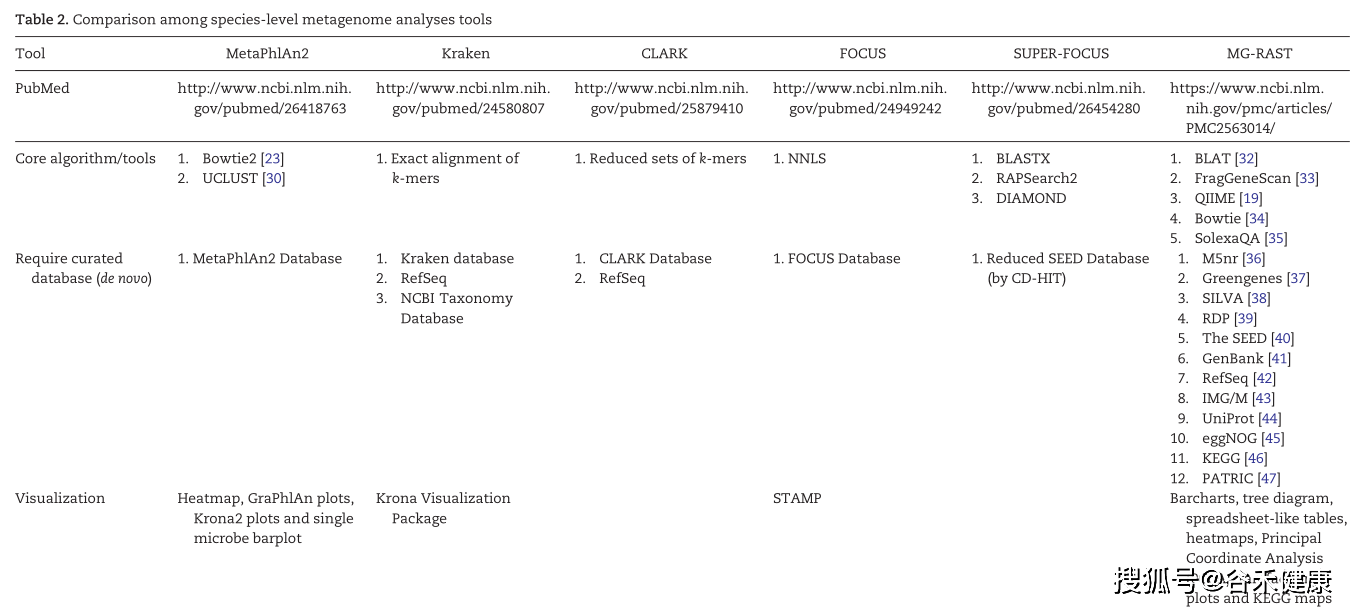
除了上面表中列出来的,另外还有
Centrifuge:比kraken2慢2x,内存使用少很多
Sourmash:类似CLARK,可以使用整个refseq作为数据库。
主流的kraken2——快速、准确度高、内存要求高
目前主要使用kraken2为主,因为快速,准确度也相当不错。不过,对于内存的要求较高,另外受数据库本身质量影响较大,默认kraken2的参考数据库只包括了细菌、古菌、病毒和人,还需要添加其他域的参考基因组。但涵盖的测序参考种仍然有限,对于菌株水平的鉴定受一定影响。后续使用Bracken可以针对kraken2的比对结果进行计算相对丰度。
MetaPhlAn2——物种跨度大、实用
MetaPhlAn2首先从全基因组数据库中找出clade-specific marker genes,然后利用这个marker genes的数据库对高通量测序得到的shotgun序列进行注释,目前主要用于后面直接使用reads获得基因和代谢通路丰度的HUMANn2的流程中,其物种跨度较大,速度也可以接受。
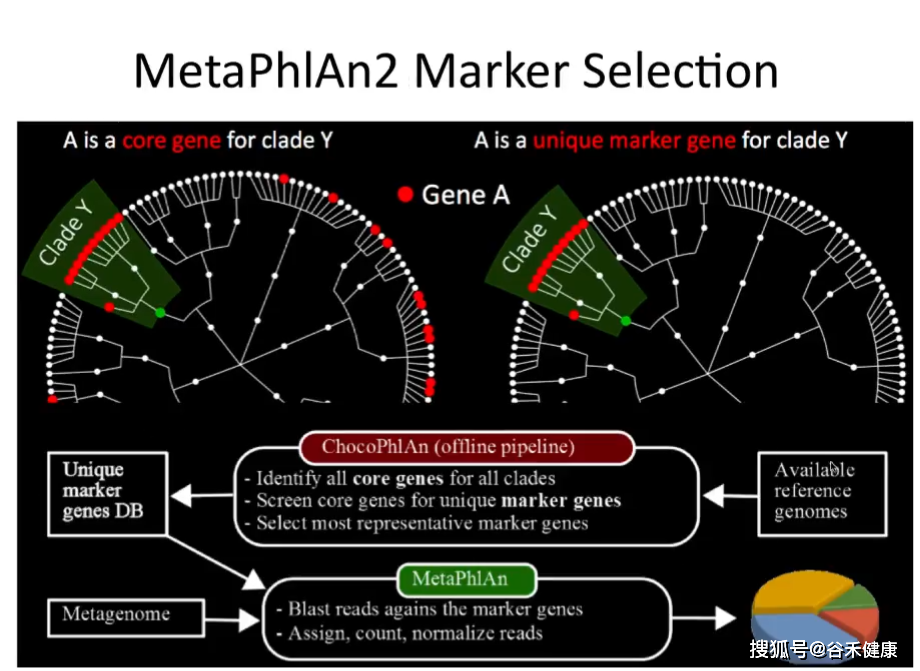
以上我们了解了直接使用reads获得丰度。
如果有足够测序深度和样本数量还可以通过组装出参考基因组来鉴定获得。该部分我们在下面的组装和分箱流程部分详细讲。
接下来,看一下我们报告中获得的结果和图:
使用Kraken2对其中的微生物进行物种注释。我们的Kraken2使用的数据库是由Refseq(2020.04.20)细菌,古细菌、真菌、原生动物和病毒库以及GRCh38人类基因组构建的。
通过查询数据库序列中的每个k-mer,然后使用所得的LCA分类单元集确定序列的适当标签,对序列进行分类。数据库中没有k-mers的序列不会被Kraken2分类。这里我们是在使用k-mer=35的条件下进行物种注释。
使用Bracken对物种注释结果计算相对丰度。Bracken是一种高度精确的统计方法,可从宏基因组学样本计算DNA序列中物种的丰度。Braken使用Kraken2分配的分类标签来估计源自样本中每种物种的读数数量。
对物种注释结果使用 KRONA 进行可视化展示。
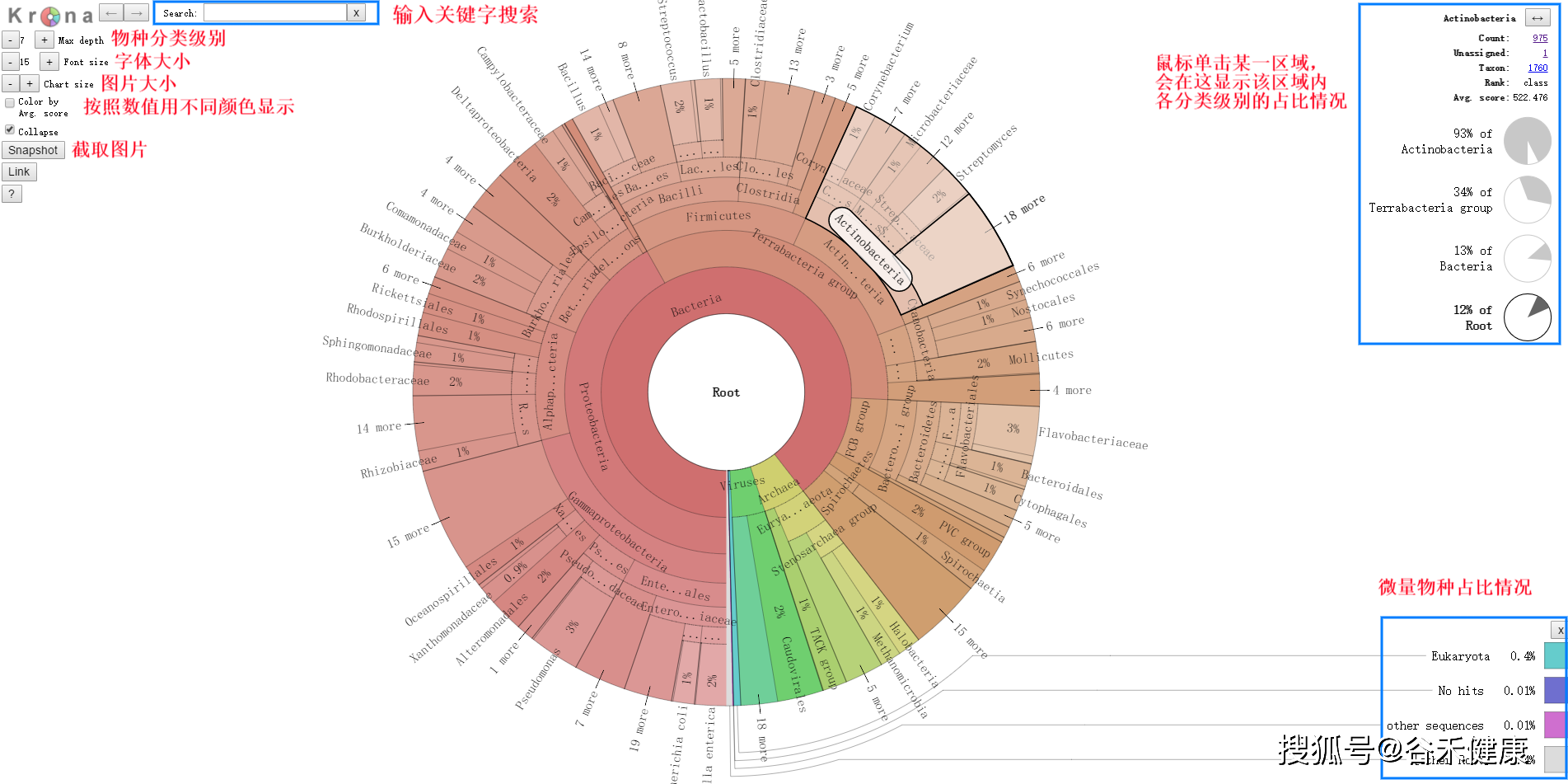
注:圆圈从内到外依次代表不同的分类级别(界门纲目科属种),扇形的大小代表不同注释结果的相对比例。
上面的是使用KRONA对单个样本的构成图形化,所有样本合并使用柱状图就可以了解具体的样本构成丰度,从门-纲-目-科-属-种-甚至菌株每个层次都可以进行显示(下面是截取我们报告中的相关图)。
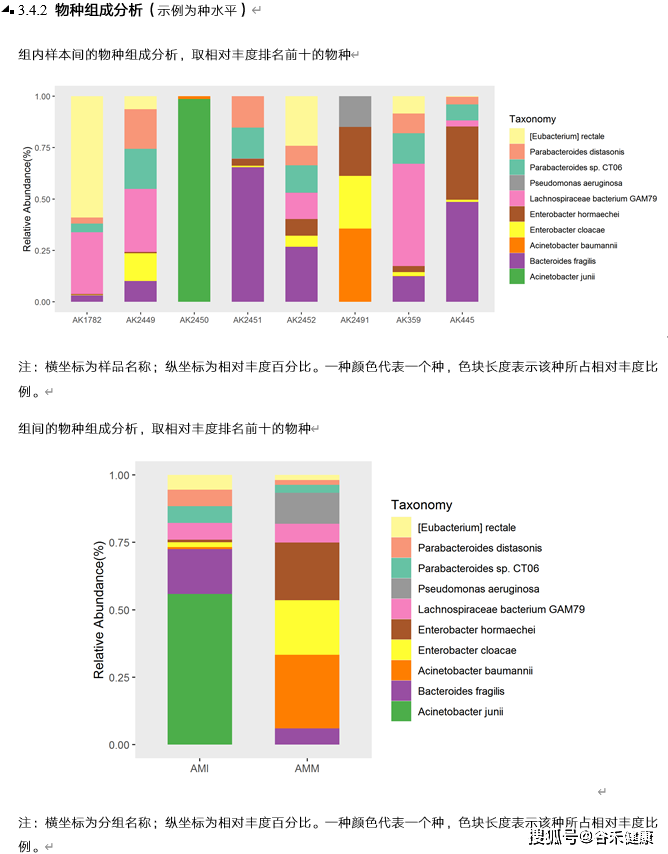
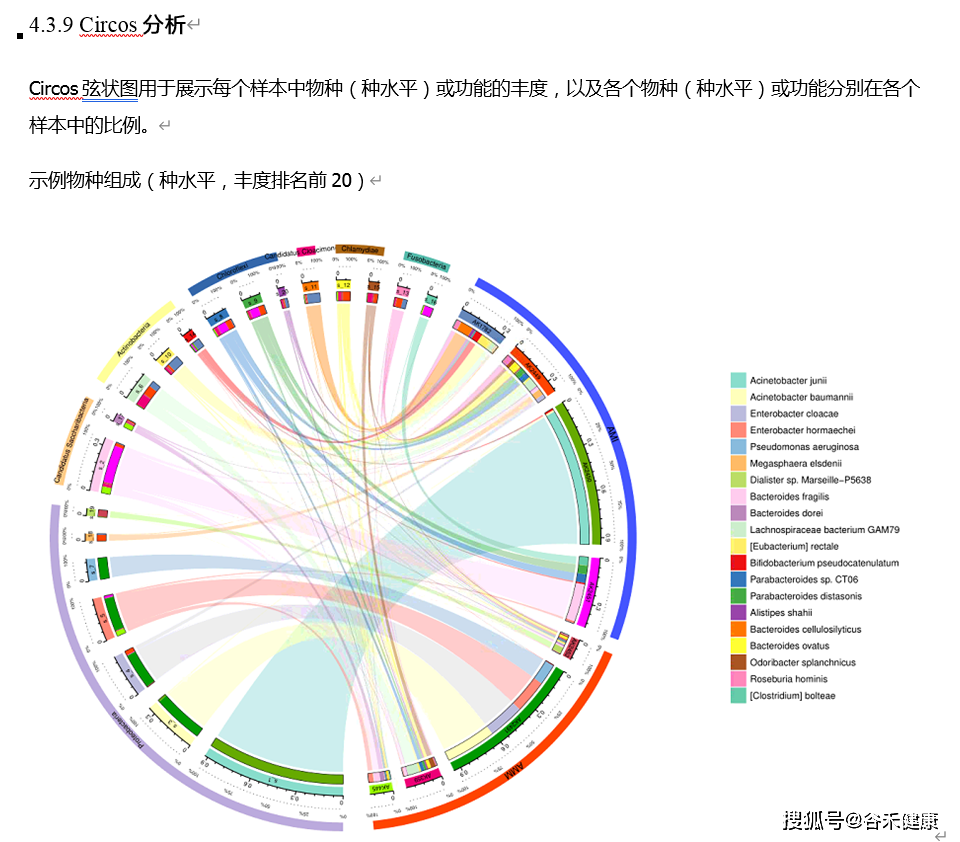
如果嫌柱状图的展示方式单一,当然也可以有别的选择。比如说以Circos的环图形式展现:
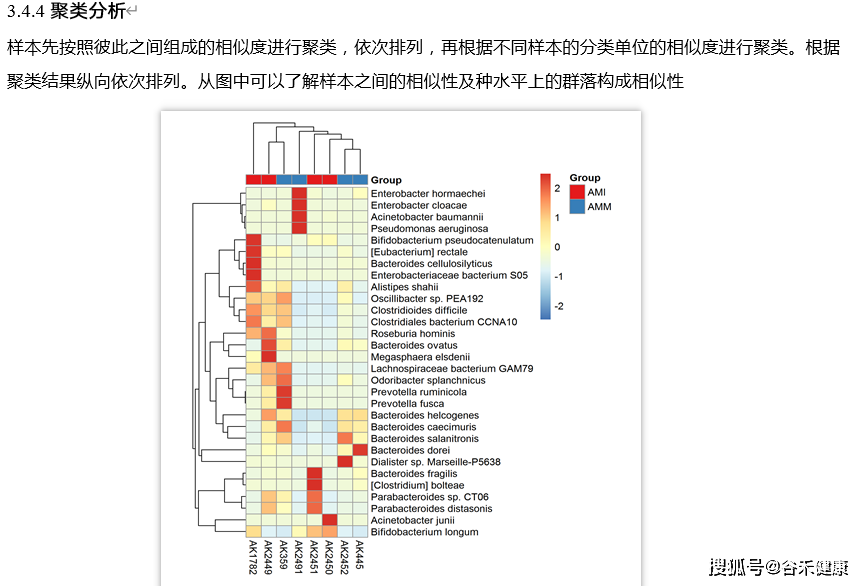
也可以进行聚类分析:
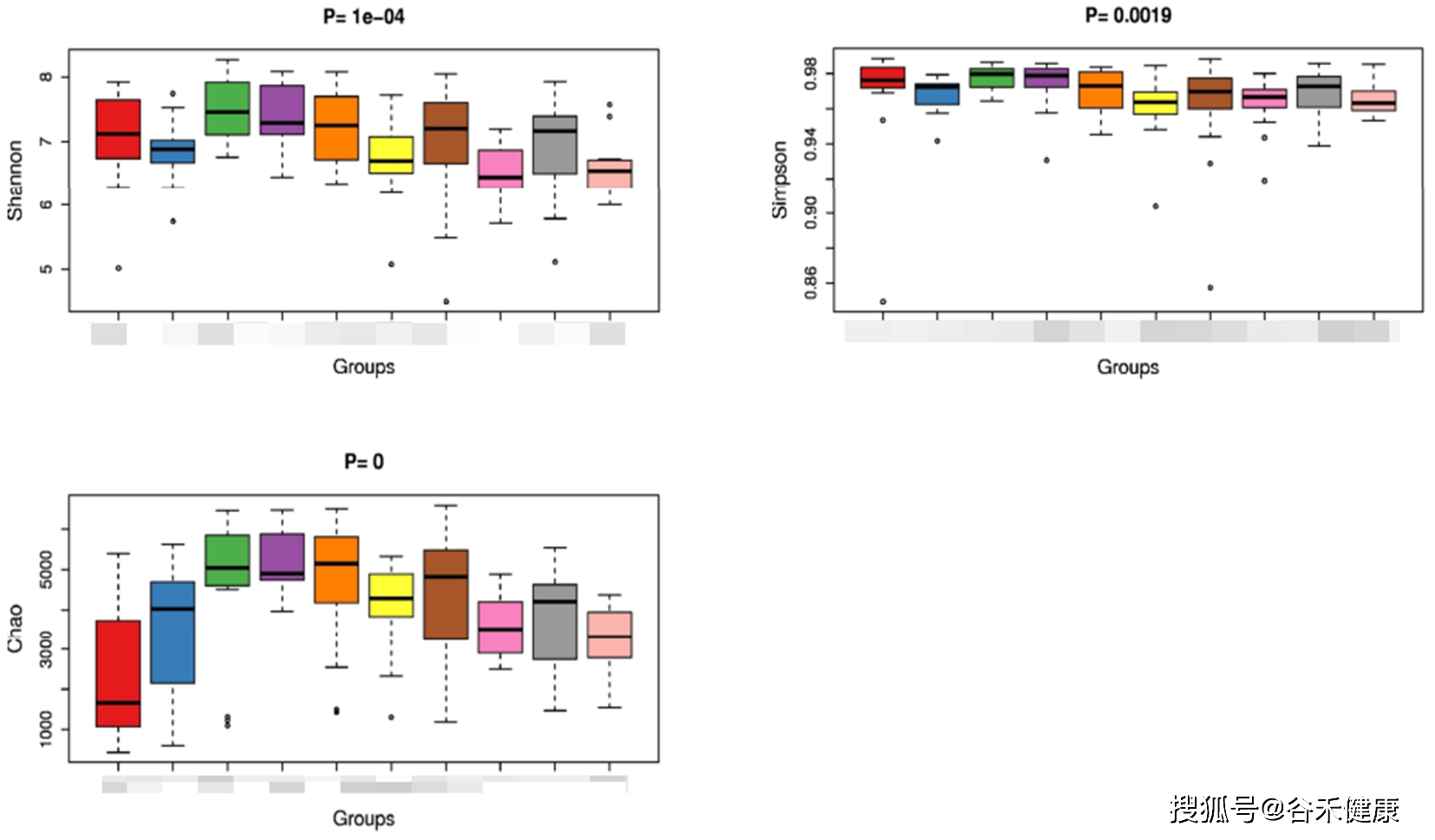
有了这些数据我们就可以进行alpha多样性(指每个样本内部菌群多样性)的分析了。
各样本和多组之间也可以进行Beta多样性的比较分析:
计算样本之间的菌属构成相似度:

组间的差异分析:寻找差异或代表性菌属,如下:

Trukey多组间检验
LefSe分析
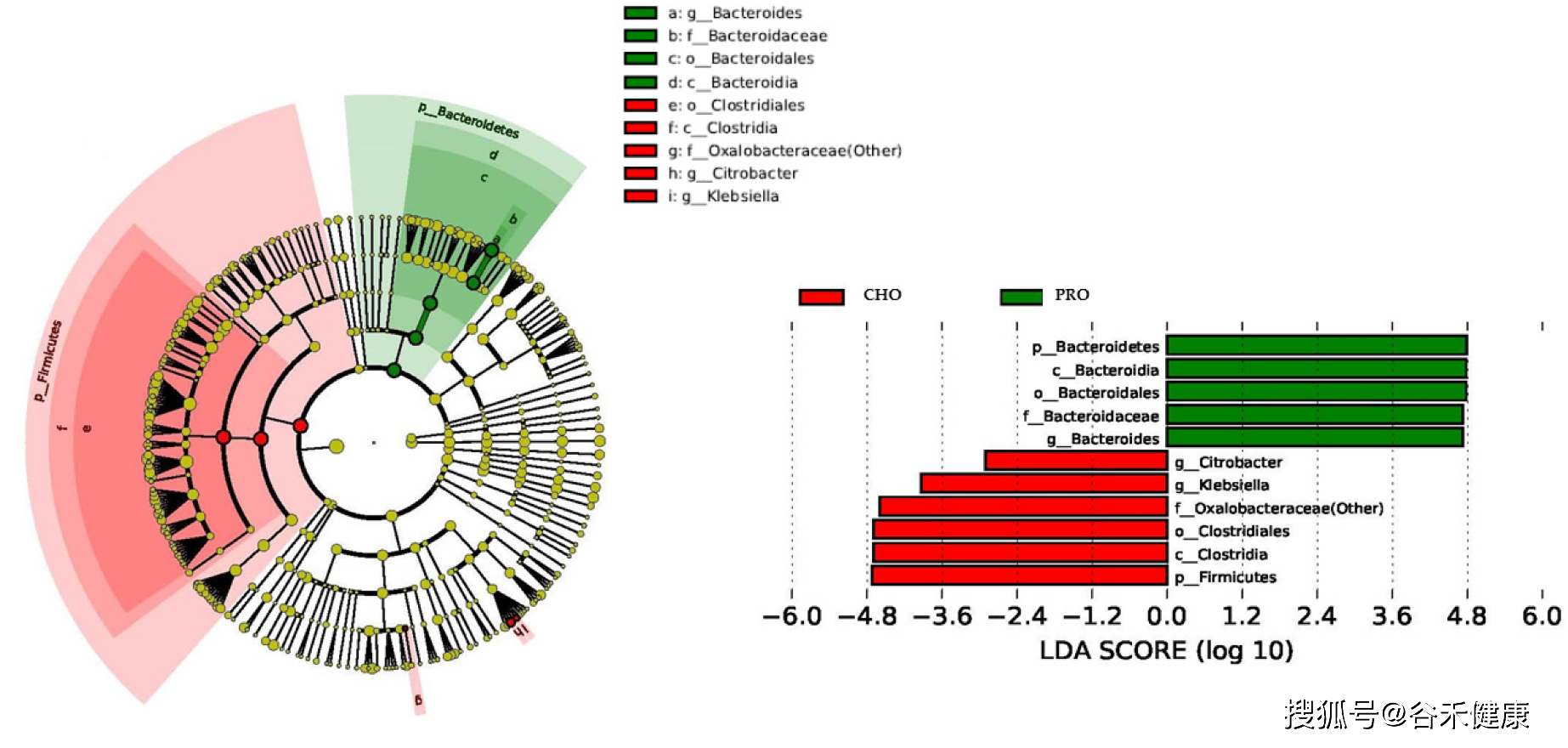
其中LEfSe基于线性判别分析(Linear discriminant analysis,LDA)的分析方法,筛选组与组之间生物标记物Biomarker(基因、通路和分类单元等),即组间差异显著物种或基因。当分组较多时较难获得每个分组独特的Biomarker。
以上是关于物种组成部分,但是有些小伙伴会有这样一些疑惑:物种构成变化很大怎么办?个体差异也很大?之类的诸多疑问。
是的,微生物群落一般对应特定的环境,其物种构成有时候变化迅速,而且个体或不同地点的构成差异极大。如人体的肠道菌群,个体之间的菌群构成差异很大,仅少量核心菌在绝大部分人的肠道内出现,个体特异性菌株也非常常见。那么如此多样性和复杂的构成如何应对相似的环境呢?
研究显示不同的菌属可能有着相似的基因或代谢能力,差异极大的种属在基因功能层面可能有着相似的构成。因此,获得微生物群的基因和功能代谢构成及分布对于解释和了解微生物如何响应和适应环境就尤为重要。
二、功能构成
下图可以帮你更好地理解上面这段话。从图中我们可以看到,舌背样本和粪便样本虽然在种属上有很大差异,但它们在基因功能层面却有着相似的构成。
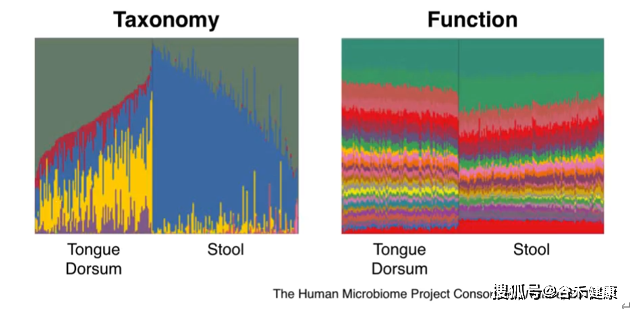
与物种构成丰度的分析类似,基因功能构成分析也同样可以包括两种方法:
方法一、通过直接基于reads的参考数据库方法获得
方法二、通过组装后预测注释基因并得到丰度
在具体展开方法之前,我们需要先了解关于基因功能的基本概念。
基因功能
每个菌的基因组中都包含大量的编码基因(ORF)以及非编码的RNA。这些基因之间又存在同源或序列相似性,达到一定相似程度的称为同源基因(一般通过CD-hit聚类为unigene,gltA这类基因名称,而数据库中一般聚类为如uniref90,eggNOG_ortholog等不同相似度的非冗余基因),这些同源基因除了序列相似同样也有着相似的功能,基于其功能或具备的蛋白功能域可以进一步分类为基因家族(Pfam),酶(EC 1.4.1.13),代谢通路(ko:K00266),更进一步层层分类为GO或顶层代谢通路Metacyc或COG等。
我们先来看方法一,具体是如何操作的?
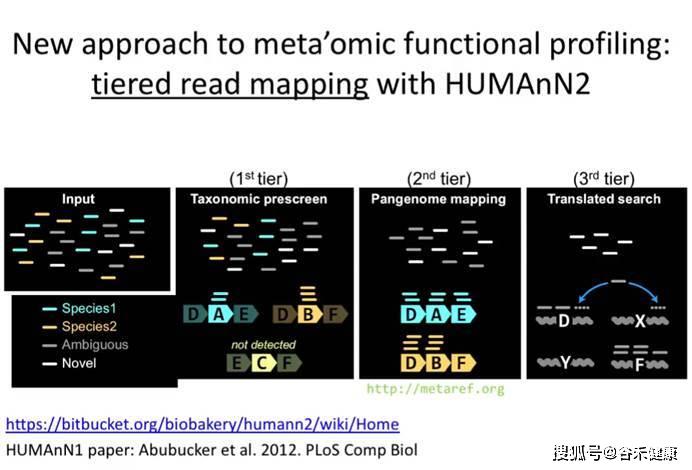
主流的HUMAnN2——获得基因和代谢通路丰度的同时可直接进行下游分析
基于测序原始序列直接获得基因构成丰度的软件目前最主要的是HUMAnN2,其首先使用MetaPhlAn2进行物种分类(关于这个软件我们在前面物种组成部分已经讲过),并提取相应物种参考基因组用于比对,未比对上的用于进一步和uniref数据库进行蛋白质序列比对。原理见下图:
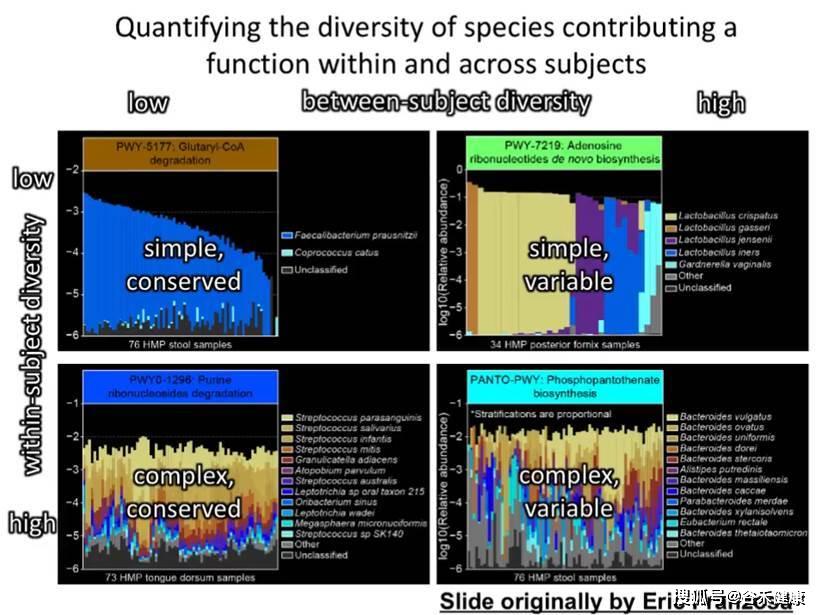
HUMAnN2的便利之处在于获得基因和代谢通路丰度的同时可以直接进行下游分析,将导出的表用于如LEFSE等差异分析,此外还可以反向给出不同样本中每个基因或代谢通路里的物种贡献。
下图是基于HUMAnN2的不同代谢通路的菌贡献比例图:
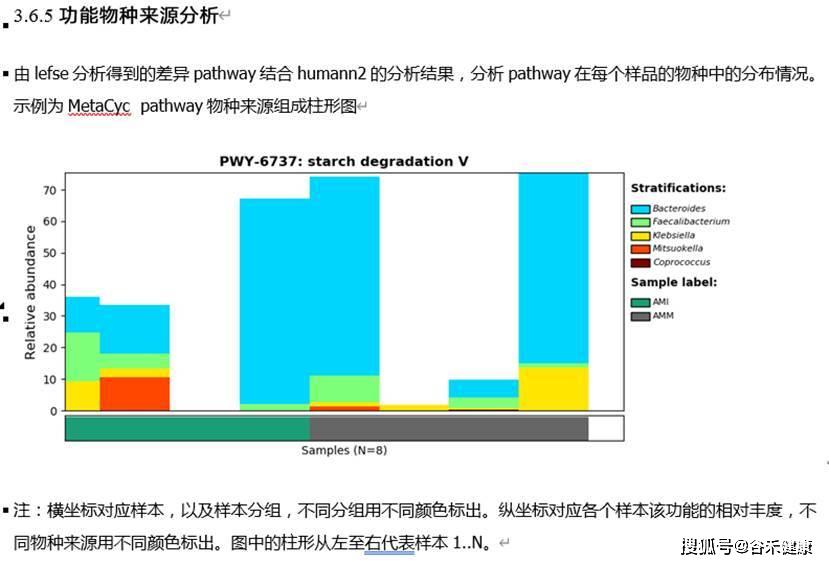
在我们的宏基因组报告中获得的是这样的:
而另外一种方法是通过组装获得,我们在前面物种构成小节也已经提到过组装分析,那么这里我们就组装拼接分析这部分展开讲解一下。
三、基于组装拼接的分析
什么样的条件下可以进行组装分析?
当测序深度足够的情况下,目前illumina二代和Pacbio以及Nanopore等长片段测序技术已经足以组装出高质量的细菌基因组草图,结合Binning方法可以一次性获得大量物种的高质量接近完成基因组。此外还有Hi-C等手段可以进一步完成基因组以及对应质粒的完整拼接。
组装的流程是什么样的?
来看一下整个基于组装的流程:
① 提取、测序
首先从样本中提取基因组DNA,进行测序,可以使用Illumina的段片段深度测序也可以辅助三代长片段测序。
② 获得contig序列
接着对序列经过质检过滤处理后直接使用序列进行拼接,获得contig序列,这时通常每个菌的基因组会有几十到数千个contig片段,由于构成复杂,很多近缘菌之间的基因组存在大量相似序列,以及每种菌丰度都不一致,所以contig阶段的片段仍然较多。
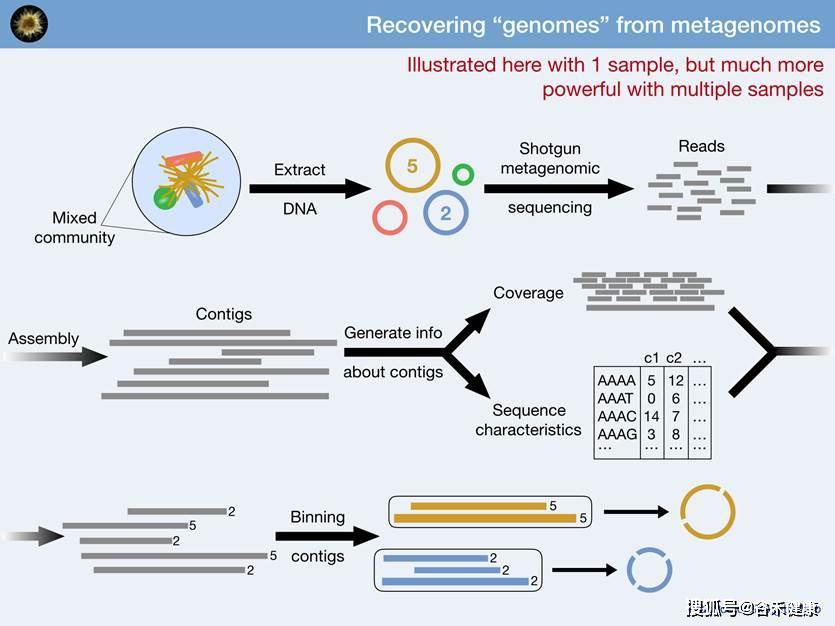
③ Binning分析
基于序列构成特征如GC含量、核苷酸多态性、覆盖度以及基因的物种相似度等多种数据,如果有多个样本或梯度可以同时结合样本丰度变化来进行分箱也就是Binning分析,将具有相同特征和变化的contig聚类归为同一个来源的箱,每个bins通常来自单一菌也就是一个菌株的基因组(我们的数据分析中包含这部分分析内容)。
④ 进一步质检评估
之后会进行进一步的质检,如checkM等评估每个Bin的完整度(核心基因以及rRNA等的完整性)和污染比例(如错误拼接,不同物种来源等)。一般要求50%以上的完整度以及10%以下的污染,当然样本数量越多,测序深度越高,测序读长越长理论上binning的质量也会更好,能获得更多高质量的单一菌完整基因组。
借用一张分箱的说明PPT:
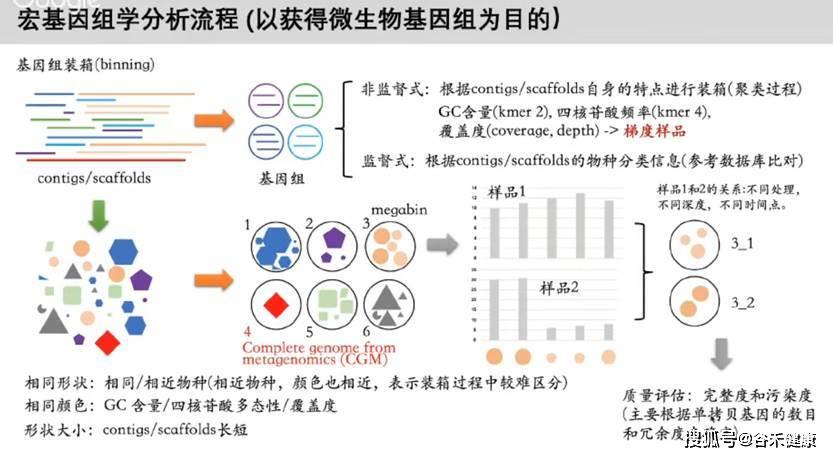
目前组装contig方面比较好的软件主要是SPAdes和MegaHIT。分箱方面MetaWRAP流程可以将整个组装和分箱优化全部完成,包括前期质检到组装以及使用三种分箱方法concoct, metabat2和maxbin2,并最终进行合并提纯优化,输出最终的分箱。
同时还可以对每个分箱bins进行物种鉴定和定量,这样我们就可以获得基于拼接组装后的物种丰度构成表,开展上述的物种多样性和样本差异统计分析。
⑤ 注释
最后使用PROKKA进行基因预测,获得的编码序列我们经过进一步CD-Hit聚类去冗余,然后使用eggNOG-mapper对其进行进一步的功能比对注释。使用salmon完成基因的定量,这样我们就得到基于组装注释的基因丰度数据了。之后就可以进行基因和功能层面的多样性、构成以及样本和组间差异分析。
我们获得的最基础的uniref,eggNOG,KEGG和GO等注释如下:
KEGG
COG
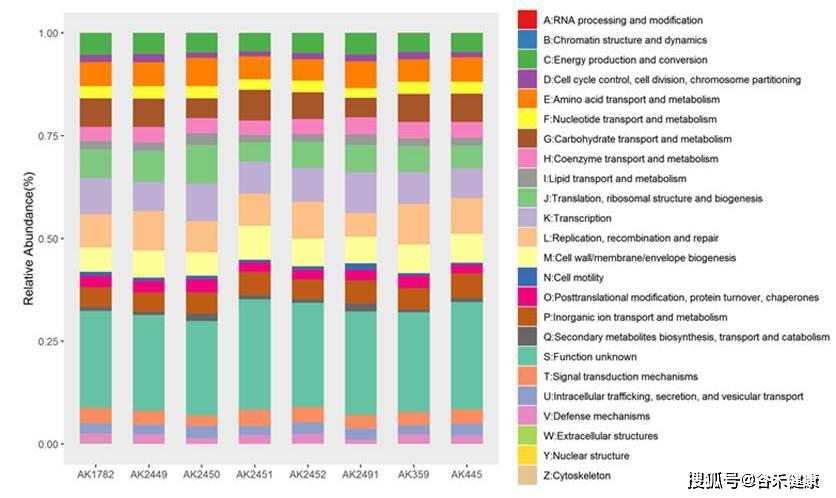
eggNOG
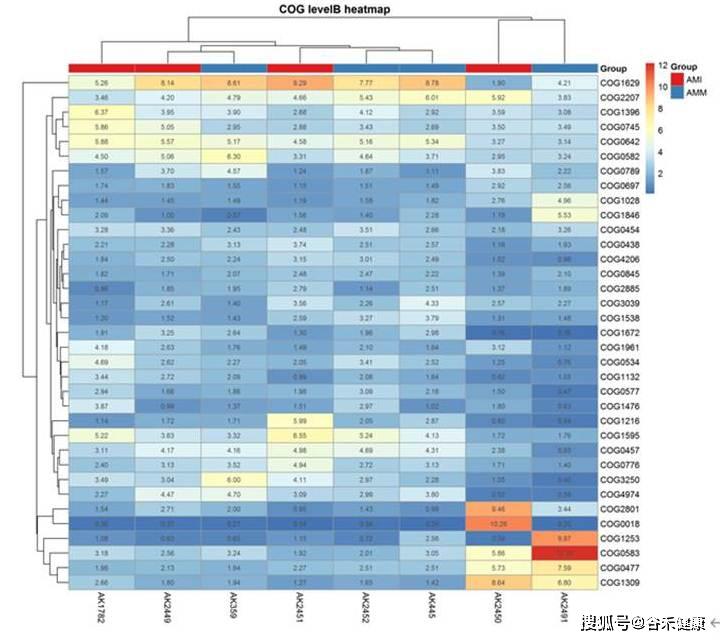
组间差异分析,如KEGG途径:
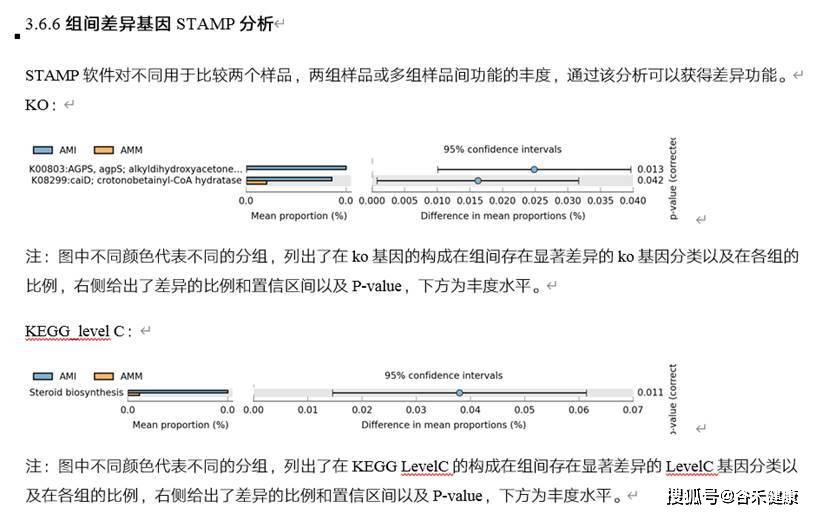
除此之外,还可以使用其他的功能基因数据库来进行进一步的基因注释和分析。比如:
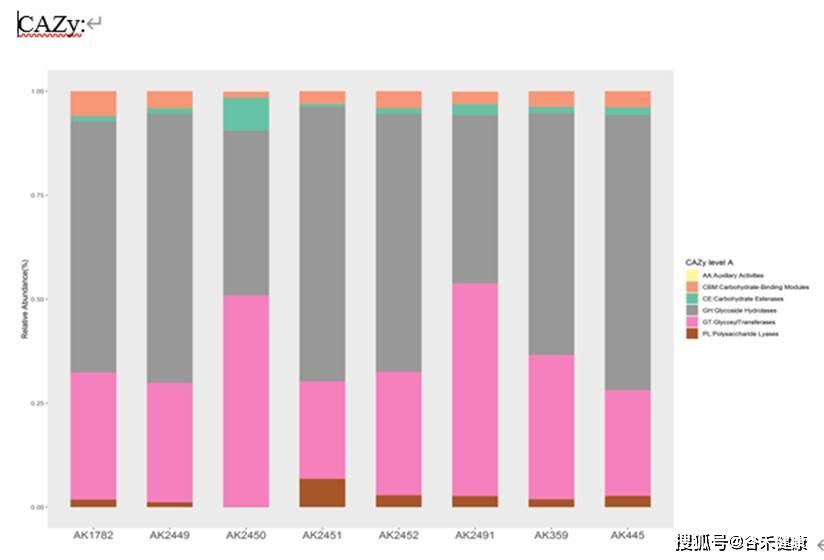
CAZy:
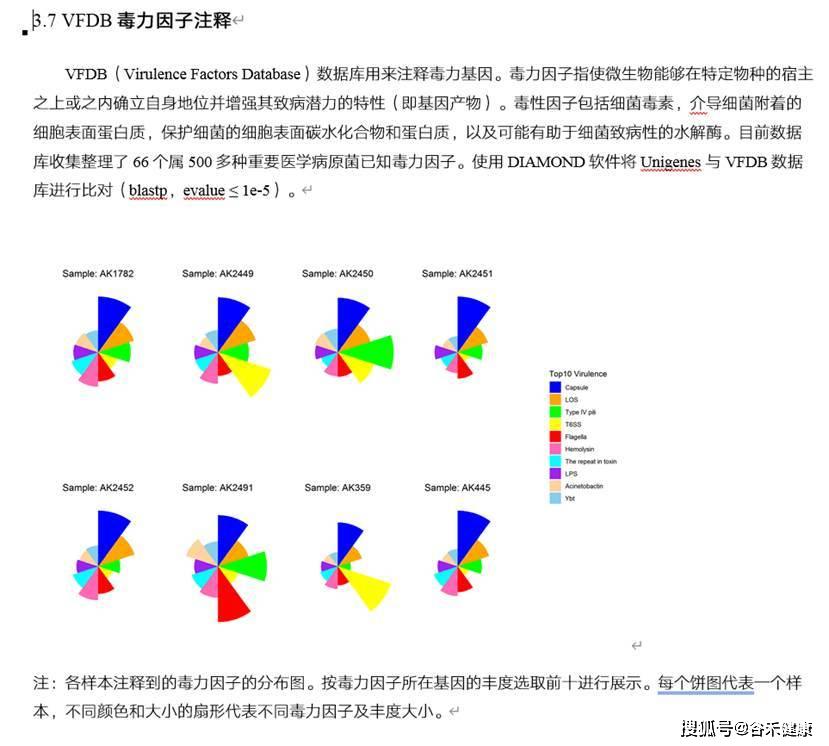
VFDB毒力因子注释:
抗性基因注释:


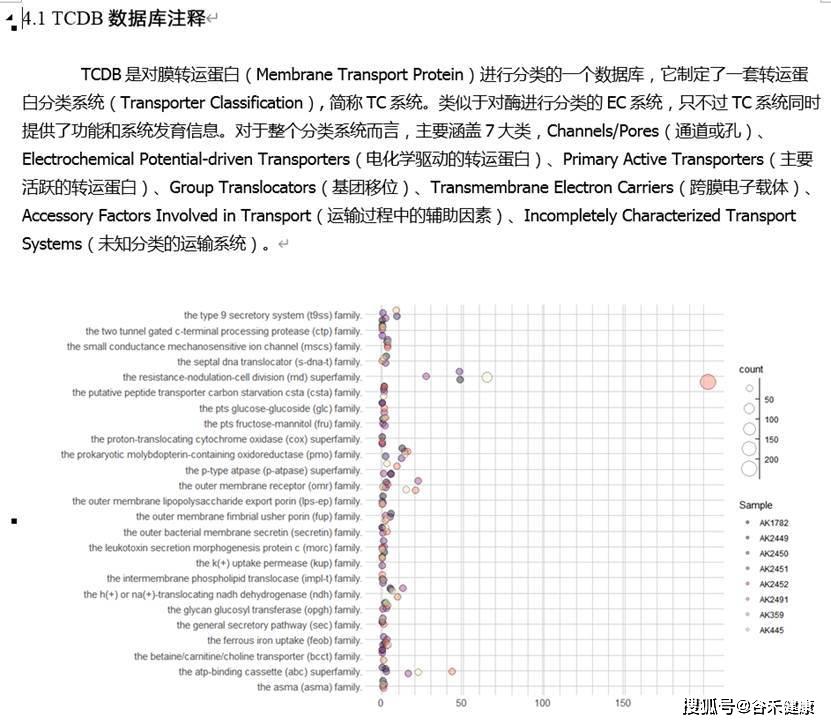
TCDB数据库注释:
PHI数据库注释:
BCGs分析:
以及基于antiSMASH和BiG-SCAPE来对代谢物的合成生物基因簇BCGs进行分析。
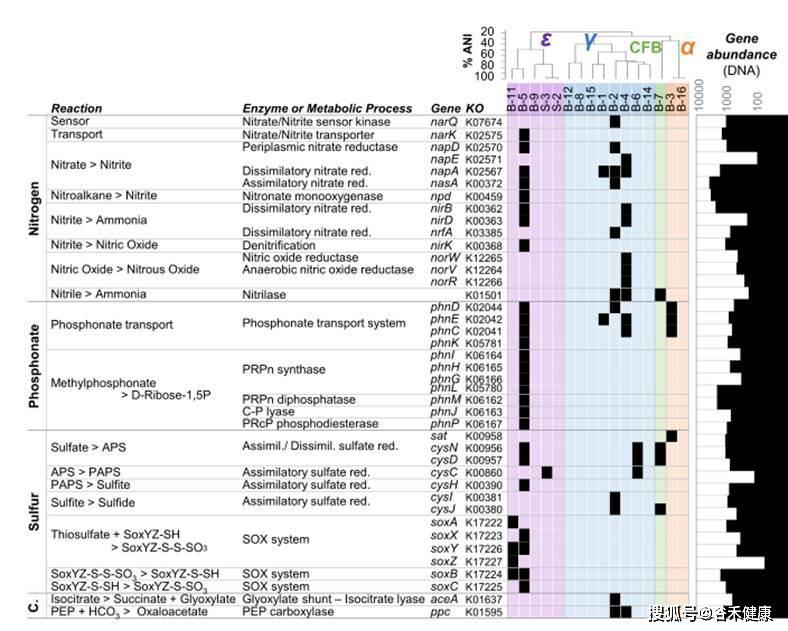
固定代谢能力评估:
或更聚焦于特定代谢的如下图中的氮、磷、硫和碳固定代谢能力和水平的评估:
当有了大量样本的菌群构成丰度信息,以及各种基因和代谢丰度数据后,我们需要根据样本的meta信息,基于不同分组,时间或环境因子等数据进行统计分析和检验,进而发现和探索可能的关联以及背后的生物学意义。
四、 统计检验
那么在面对宏基因组这类数据时在进行统计检验分析时需要注意什么呢,应该采用哪些分析,并如何解读这些结果呢?
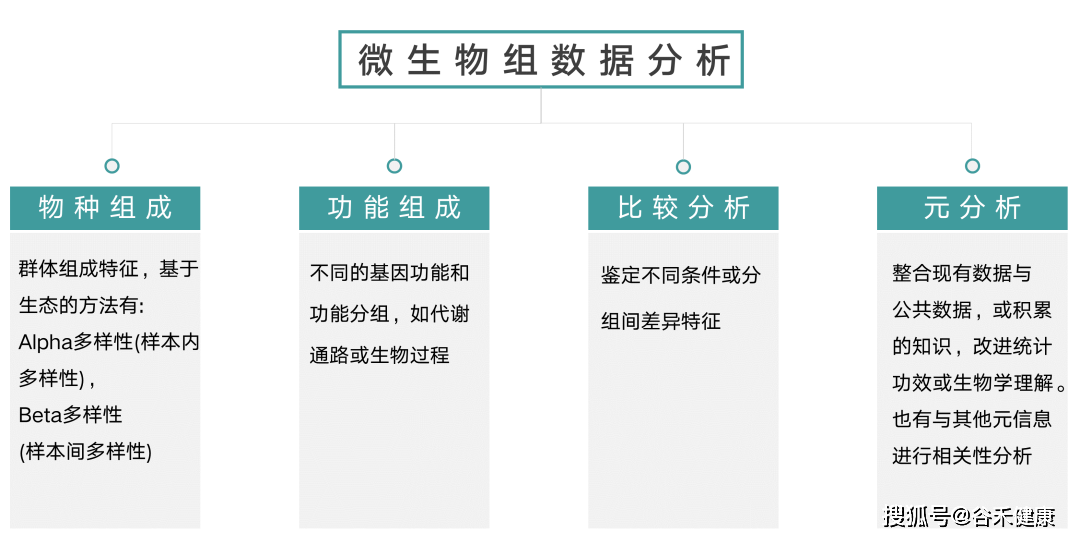
首先,微生物组数据分析分为四大类:
在对所有数据进行统计检验前一般建议对数据进行基本的质量过滤。一类是去除绝大部分样本都不存在的物种和基因,如Prevalence in samples (20%),还有一类是去除变异度过小的Percentage to remove (10%)基于Inter-quantile range。
为什么可以过滤这两类?
上述的两类由于其携带的信息量和变化过小在进行组间比较统计检验的时候都建议过滤,因为要么是污染,要么与差异无关。
宏基因组数据具有一些独特的特征,例如测序深度的巨大差异,稀疏性(包含许多零)和分布的巨大差异(过度分散)。在进行后续的统计检验之前建议针对不同的分析方法进行相应匹配的标准化处理。标准化包括:
Rarefaction和缩放方法:这些方法通过将样本放到相同的比例进行比较来处理不均匀的测序深度;
转换方法:包括处理稀疏性,组成性和数据中较大变化的方法。
那么各种标准化方法是什么,应该选择哪种方法?
参考MicrobiomeAnalyst网站提供的信息,以下是一个简短的介绍:

请注意,数据标准化主要用于可视数据探索,例如beta多样性和聚类分析。有时候不使用标准化也能获得最佳结果,比如:单变量统计和LEfSe。
同时,其他比较分析将使用其自己的特定标准化方法。例如,对metagenomeSeq使用累积总和定标(CSS)标准化,对edgeR应用M值的修剪均值(TMM)。
经常有小伙伴问,这个数据是用的什么标准化?没有做标准化怎么办?这类问题。
目前,尚无关于应使用标准化的共识性指南。建议大家可以探索不同的方法,然后目视检查分离模式(即PCoA图)以评估不同标准化程序对实验条件或其他感兴趣的宏基因组数据的影响。
有关这些方法的详细讨论,请参考使用者最近发表的两篇论文
① Paul J. McMurdie等
(https://doi.org/10.1371/journal.pcbi.1003531)
② Jonathan Thorsen等
( http://doi.org/10.1186/s40168-016-0208-8 )
以上是关于标准化的这部分内容需要了解的知识,接下来我们来看具体如何操作,怎么得到那些图表?它们分别代表什么?
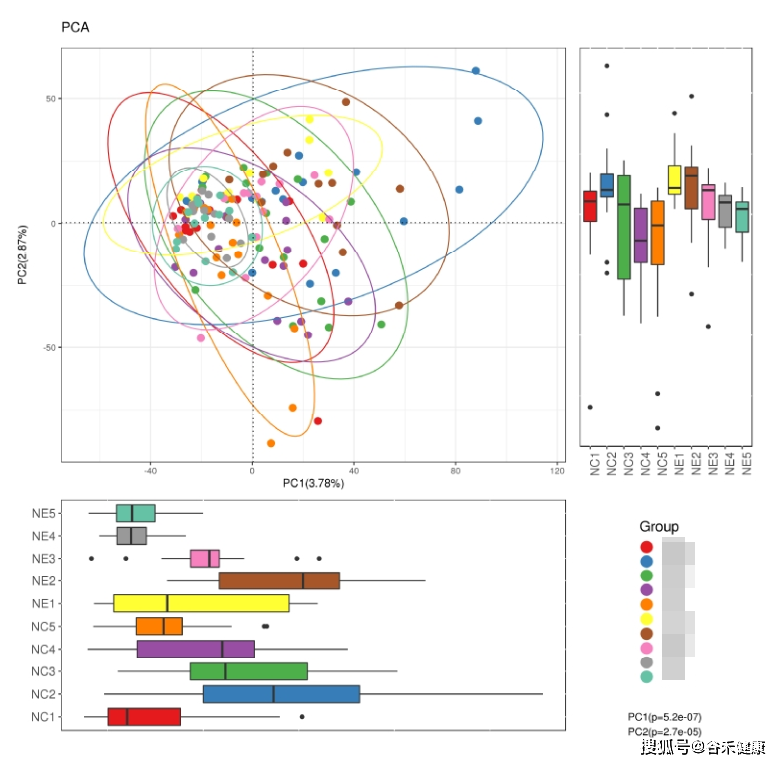
一般我们需要先进行探索性分析,也就是不设预订的假设,首先从主成分分析结果中了解样本的菌属和基因的大概分布。
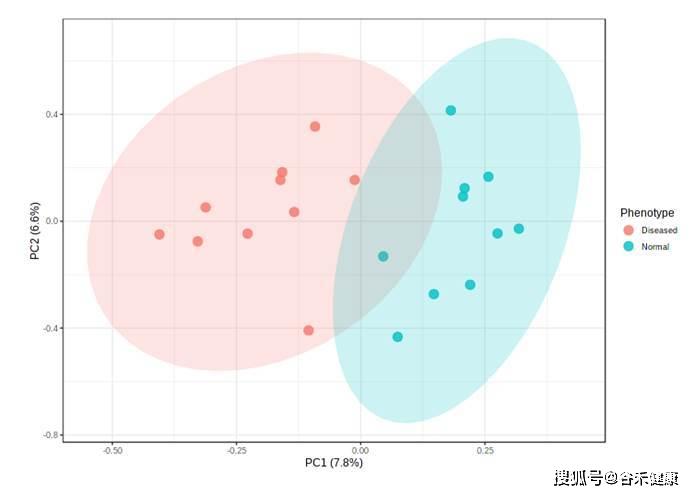
主成分分析是根据不同距离算法计算样本之间的距离矩阵,然后进行降维,最终形成一个三维的空间分布。样本之间在空间上分隔越远表明样本之间的差异越大。
比如我们报告中的下图,疾病和正常样本可以较好的区分,一般此处我们还会进行一个统计检验,来判别PC1和PC2这几个维度上两组之间是否真的存在统计差异。
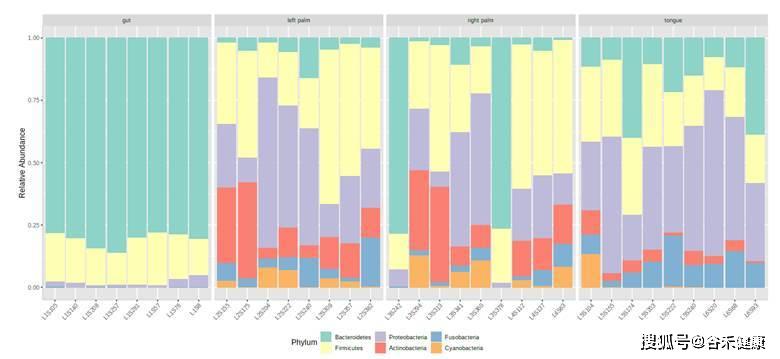
基于丰度图来评估各样本和分组的基本构成,如:
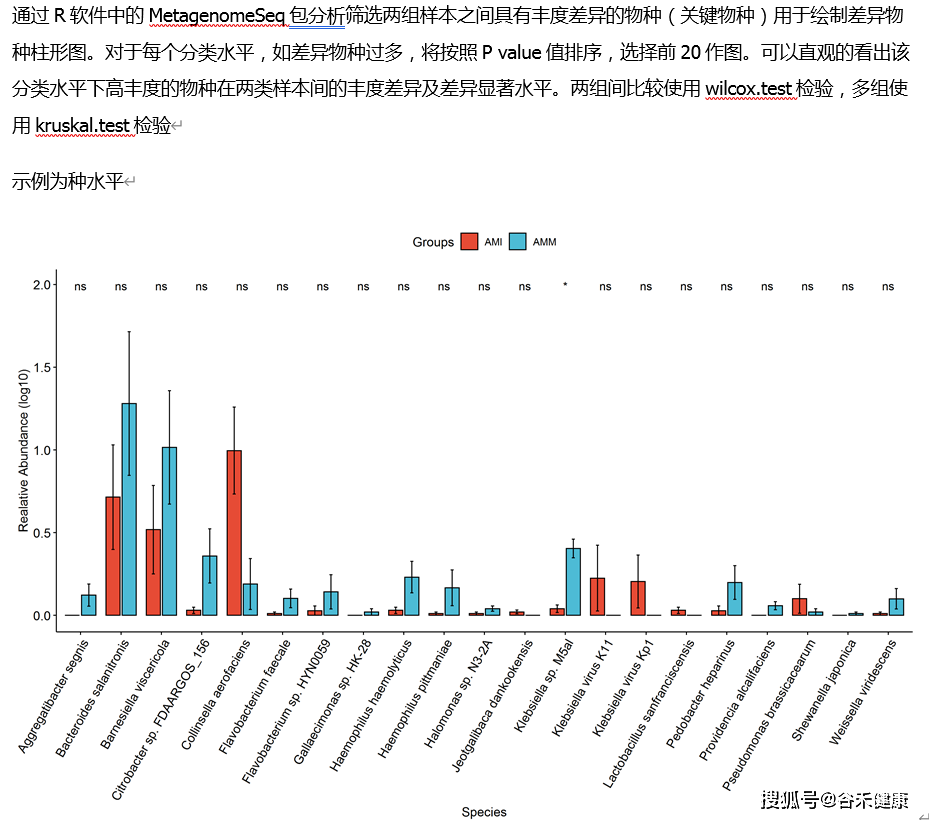
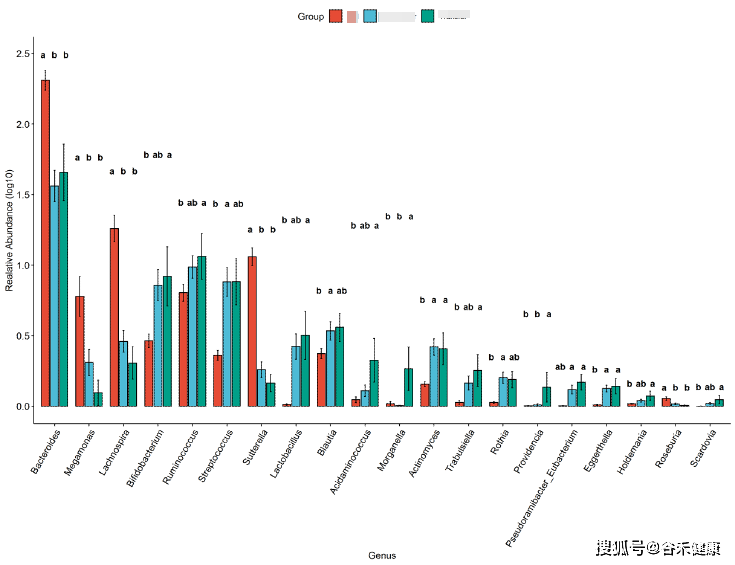
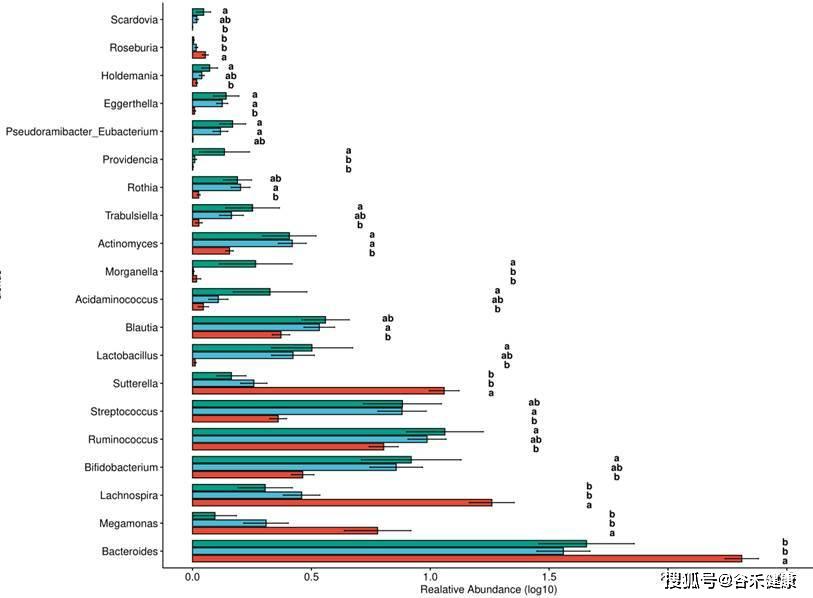
之后我们可以针对不同分组或处理之间的样本进行统计检验,可以使用的检验方法包括两组间的非参数统计检验T-test/ANOVA,3组以上组间统计检验可以使用Tukey test,其直接生成各组将的统计差异,并提供字母标注,直观简便,如:
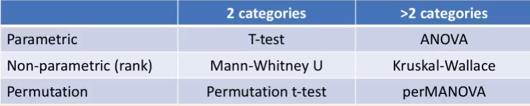
具体的统计方法选择可以参考下表:
除了常规的非参数检验外,包括metagenomeSeq和DEseq以及edgeR等统计方法包可以很好的分析组间差异特征。LEfSe则一般用于寻找特征标志物。
那么有了大量的差异特征菌属或基因之后,我们是否能基于这些差异菌属有效的区分不同的分组呢,或构件一个模型来预测或分类呢?
这时候可以使用随机森林(Random Forest)一类的决策树机器学习模型,来利用这些差异特征构建分类模型,并使用AUC等指标来评估基于这些模型的预测有效性和准确度(我们报告中如下图)。
当然也可以使用其他更复杂的如深度学习等方法来构建分类模型。
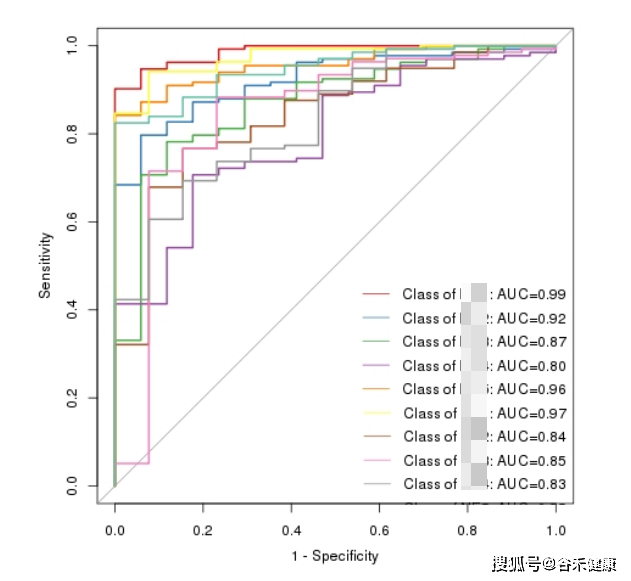
除了性别、疾病、地点等分类差异之外,我们通常还有很多元数据,包括临床指标或环境因子等信息,这些数据通常是连续型数值,对于这类数据我们可以进行相关性分析。
当然反过来,将菌群特征作为表型也可以和如基因组的基因型或SNP构成来进行相关性分析。
对于菌群数据的相关性分析比较推荐:
SparCC方法,可以构建菌种或菌属之间的相关性网络,相对稳定。
对于与疾病或环境变量进行相关性分析可以使用:
Sperman秩相关分析。
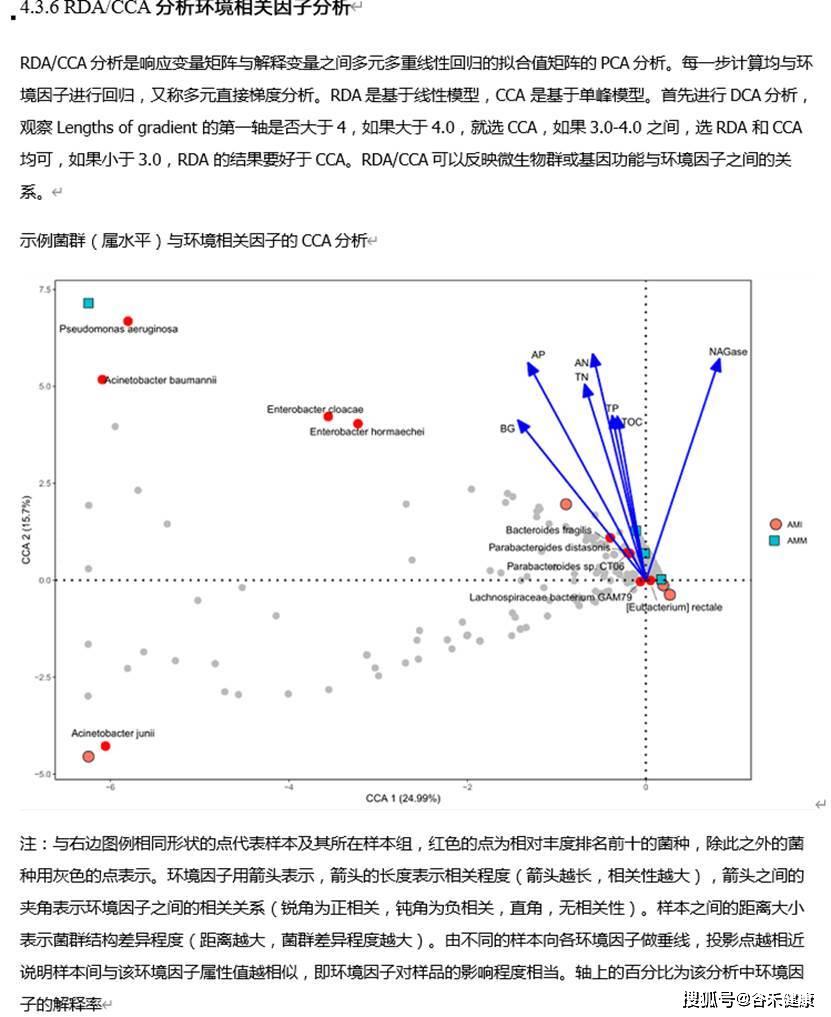
另外RDA/CCA分析也可以有效的反映菌属与环境因子等指标直接的关系(我们报告中如下图)。
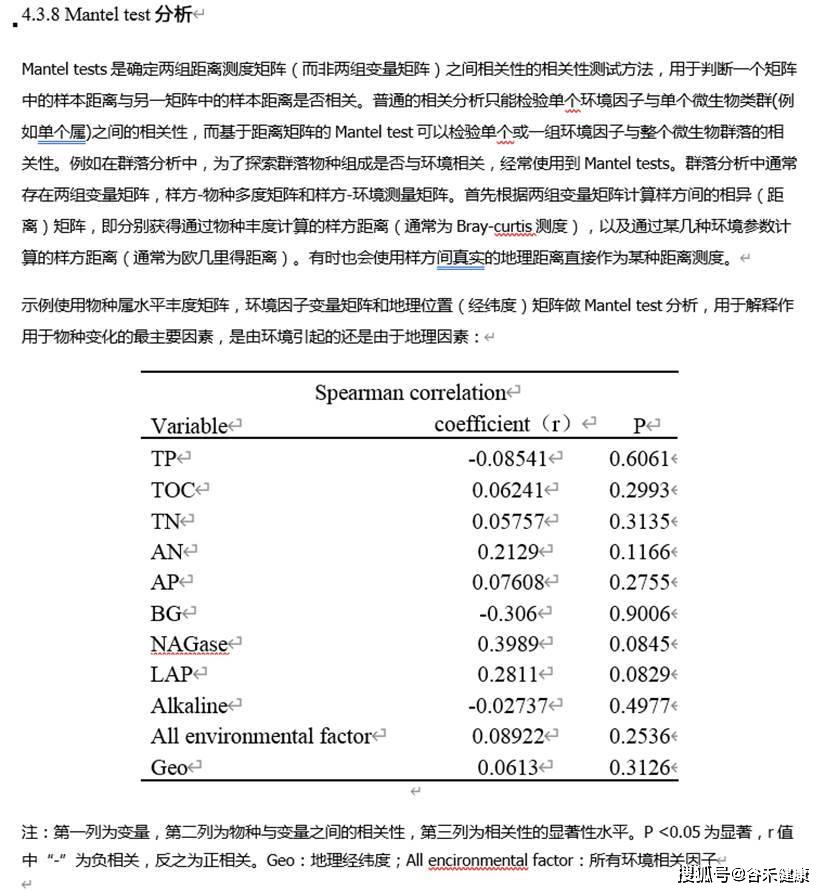
Mentel检验也可以用于判断菌群构成特征与单个或一组环境因子之间是否存在显著相关。
要 点
宏基因组从大量菌群和基因构成中寻找关联是需要足够的样本量才能达到有效的统计效力,因为一次性获得了大量的特征数据,样本量过少会带来统计结论的无效,越是组内差异大,组间差异小的研究足够大的样本量才能得到可靠的结论。
一般动物样本具有较好的背景可控,组内样本数量建议至少6个,而人群研究由于背景复杂,个体多样性高,一般建议组内50例以上较好。
以上看完后,你应该对宏基因组的数据分析流程有了整体的认识,也学会了相应的一些操作,但是不一定能直接从自己的这些数据、图表中真正探索到和实际生物学相关的有价值的研究成果。
所以,我们又选取了一些已发表的研究作为案例,结合实际问题来具体分析,从实验设计到具体分析流程方法和图表的展示,再到相应的结论,掌握这类文章的总体思路。
之后无论是刚开始的实验设计,还是后面的分析,都会更加得心应手。
建议想好整个实验思路再开始(或者也可以咨询我们,我们专业的数据分析团队会为你提供切实可行的项目方案)。
04
案例解析
< 案例一 > 肥胖患者的肠道微生物组
第一项研究是关于肥胖患者减肥手术后的宏基因组和代谢数据的分析研究。
文献来源:Aron-Wisnewsky J, Prifti E, Belda E, et al. Major microbiota dysbiosis in severe obesity: fate after bariatric surgery. Gut. 2019;68(1):70–82. doi:10.1136/gutjnl-2018-316103
研究纳入了61名严重肥胖的受试者,他们是可调节胃束带术(AGB,n = 20)或Roux-en-Y胃旁路术(RYGB,n = 41)的候选人。减肥手术后1、3和12个月随访24名受试者。使用宏基因组学测序和LC-MS分析肠道菌群和血清代谢组。另外纳入了10人和147人分别作为宏基因组和代谢检测的验证集。
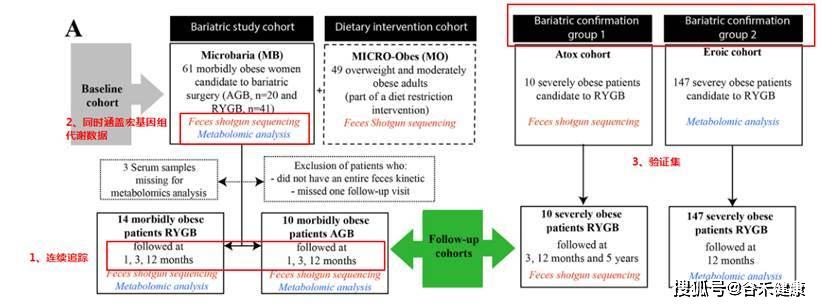
研究思路
这样的设计分别有什么作用?
第一点持续的动态采样可以获得持续变化情况,尤其是在一个特定变化后(减肥手术),持续的最终采样有助于确认菌群的变化出现和特定事件或生理病理变化的前后,尤其是在确定因果中有重要帮助。
第二点获得多维的数据有助于帮助我们全方位的了解菌群变化背后的带来的生理和代谢变化以及之间的关联。
第三点独立验证集的存在将大大增强研究的可信度,尤其是该研究纳入的样本量并不多,无法全面有效的控制无关因素,使得很多统计检验的效力无法显现。这也导致该研究仅在基因总量和多样性上获得较好的重复效果,而更多的菌群精细特征以及具体基因和代谢通路没有得到深入分析。但是独立验证集保证了核心结论的可靠性和重复性,这点在宏基因组研究中非常重要。
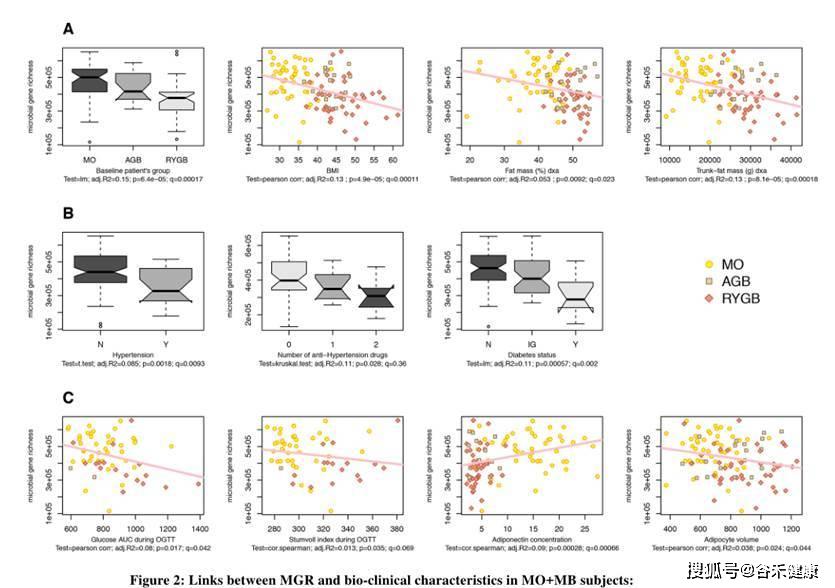
从下图可以看到研究针对样本的总基因多样性水平与生理指标和疾病状态进行相关性分析和组间差异分析,图中给出了显著相关和差异的指标。
使用的统计检验方法是pearson和sperman相关和t-test以及Kruskal-Wallis检验。
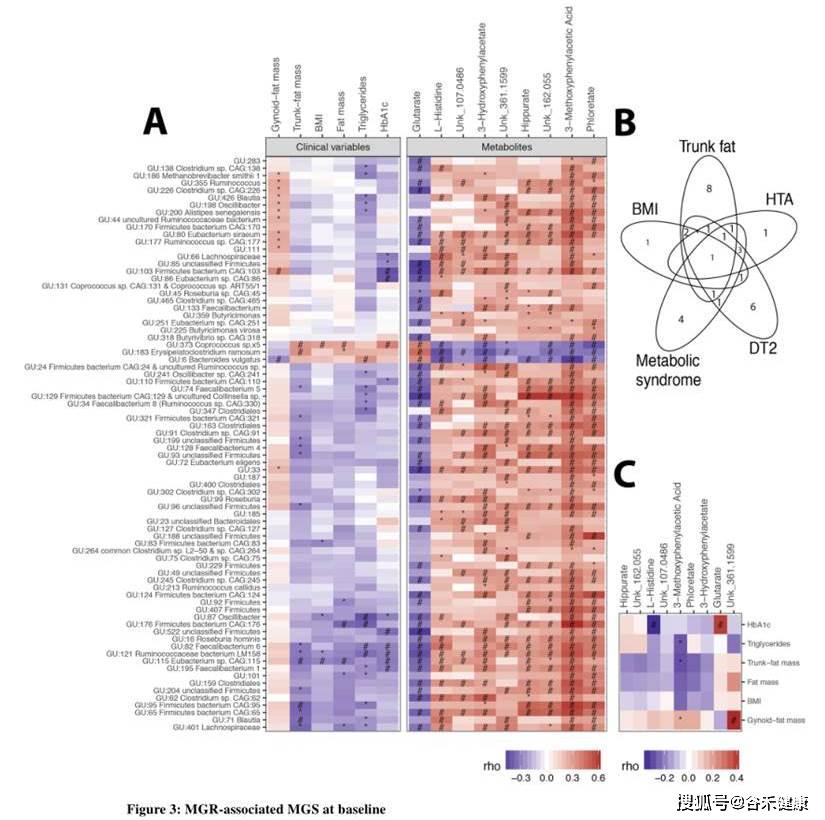
下图是研究将MAGs与各项生理和代谢值进行相关性分析后的热力图。该研究由于测序较早,并未独立拼接,而是直接使用了之前一项人类肠道菌群研究获得组装基因组参考序列。
进一步研究分析了术后特定变化模式的MAGs以及它们与代谢生理指标的相关性,见下图:
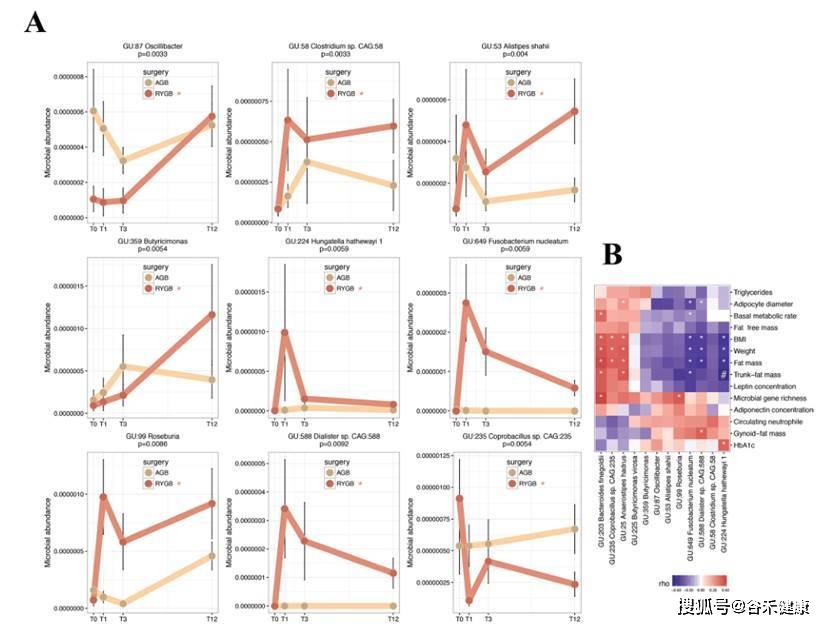
上图的研究可以通过pattern search的方法寻找特定变化模式的菌种。
研究的主要结论发现是低基因丰富度(LGC)存在于75%的患者中,并且与躯干脂肪质量和合并症(2型糖尿病,高血压和严重程度)增加相关。LGC改变了78种宏基因组种(MGS),其中50%与不良的身体成分和代谢表型有关。九种血清代谢产物(包括谷氨酸盐,3-甲氧基苯基乙酸和L-组氨酸)和含有参与其代谢的蛋白质家族的功能模块与低MGR密切相关。术后一年,BS会增加MGR,但尽管RYGB患者的代谢改善比AGB患者大,但术后一年的MGR仍然很低。
点 评
总体而言该项研究可以使用浅宏基因组(在文章开头第二部分详细介绍过)来完成所有测序和分析,进一步扩大样本数量,如果能同时获得人的转录组数据甚至能更加明确的找到菌群变化与特定代谢通路的关联关系。
< 案例二 > 食物与人类肠道微生物组
第二项研究是Dan Knights实验室发表在Cell Host & Microbe,2019的一篇针对34个人17天每日饮食和菌群变化的相关研究,试图揭示日常食物选择与人类肠道微生物组组成之间的精细关系。
文献来源:Johnson Abigail J,Vangay Pajau,Al-Ghalith Gabriel A et al. Daily Sampling Reveals Personalized Diet-Microbiome Associations in Humans.[J] .Cell Host Microbe, 2019, 25: 789-802.e5.
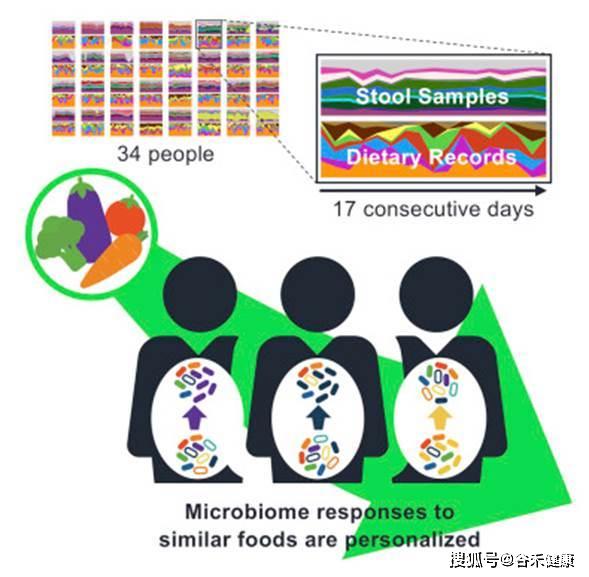
可以看到,研究同时记录了粪便样本的菌群宏基因组和每日的饮食记录。研究的核心在于将每日饮食的食物通过营养构成进行量化,并构建类似物种进化树的食物物候树。
此外由于有每日的数据,可以通过前一日的食物与第二日的菌群数据进行时间序列分析,构建食物与菌之间的关联以及时间相关性。
最后基于菌群数据和前一日饮食来构建模型预测判断后一日的菌群状态,帮助我们了解食物对于个体菌群的影响因素并实现定量和预测。
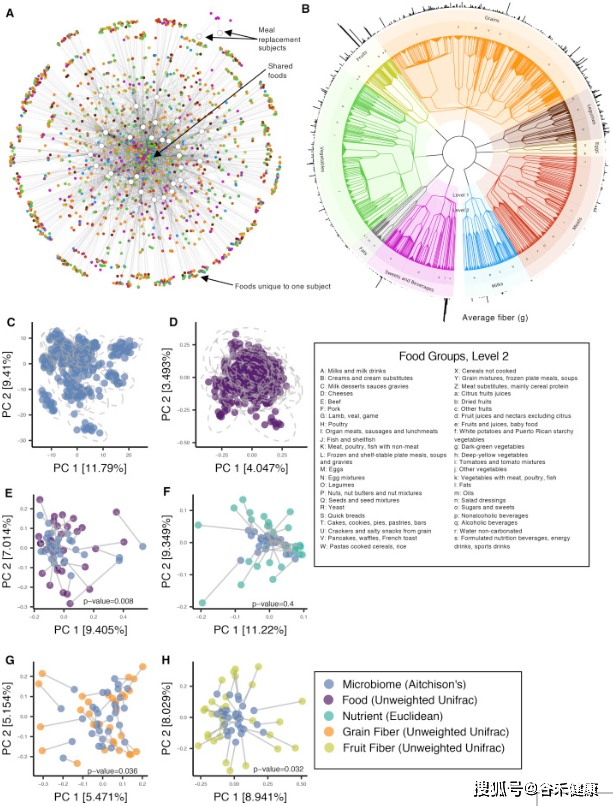
研究中对数据的处理过滤标准如下:删除所有具有低读取计数(每个样品<23,500个读取)的样品。物种级别的分类表仅限于研究对象中至少存在25%的研究日,且在10%的研究样本对象中发现的那些物种。
最后,相对丰度<0.01%的稀有物种被丢弃,将物种数量限制为290个注释。将得到的分类表汇总到较高的分类级别(即属,科,门等),以进行下游分析。
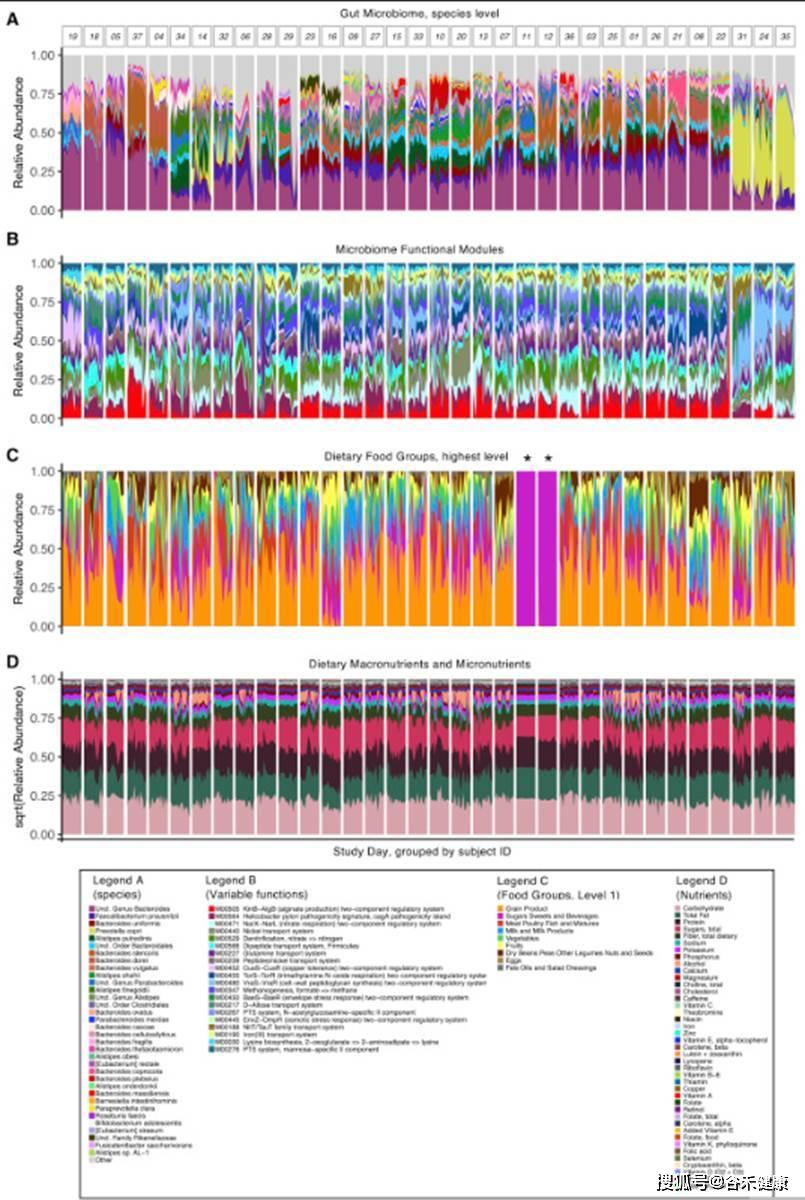
菌群和饮食以及营养构成的堆叠图很好展现了变化和对应。
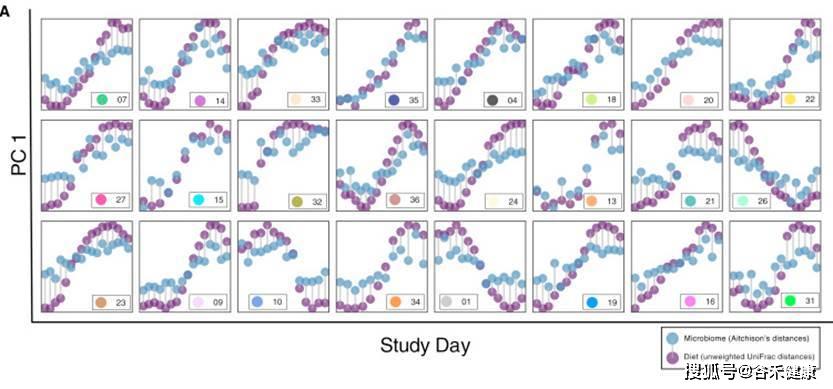
下面这张图很好的显示了饮食食物的变化与菌群变化之间的时间变化关系:
下面这张图通过对每个人单独进行菌属与食物的Spearman相关,展现了菌与食物之间的关联的个体化差异,在特定菌属对应相同食物不同人会出现完全不同方向的变化,这也正是这项研究所揭示的,这种关联关系的复杂性。
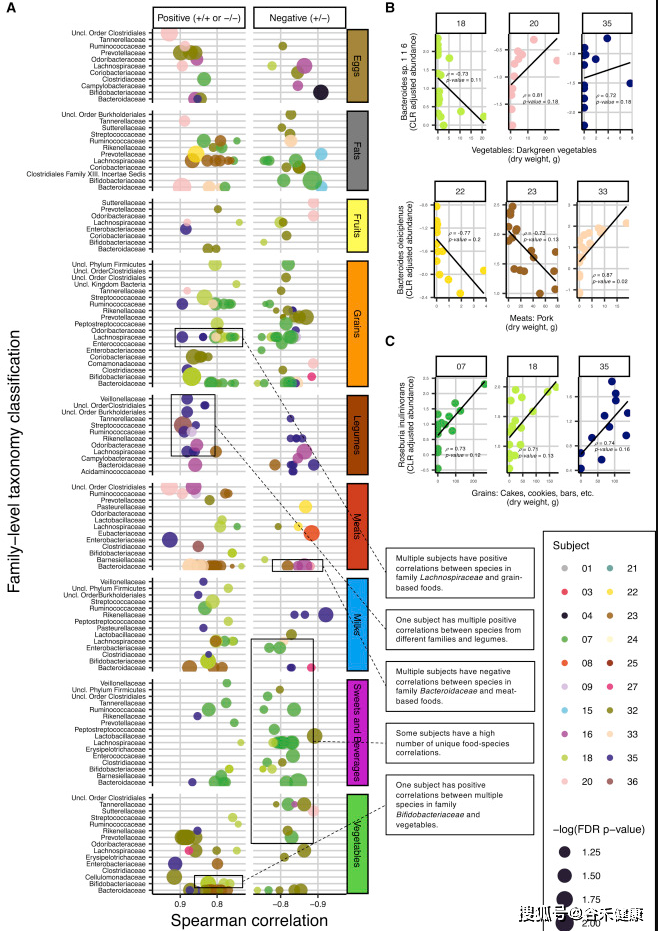
点 评
本研究虽然有大量样本,但并未进行组装,而是直接使用了Refseq的细菌完成基因组序列作为参考。研究由于样本数量众多,测序深度也很有限,类似研究也可以使用浅宏基因组方式完成。
< 案例三 > 类风湿关节炎的人群肠道微生物组
接下来的一个研究是比较典型的宏基因组组装并与疾病进行关联分析的案例,研究的是日本人群类风湿关节炎的肠道微生物组的全基因组关联研究。
文献来源:Kishikawa Toshihiro, Maeda Yuichi,Nii Takuro et al. Metagenome-wide association study of gut microbiome revealed novel aetiology of rheumatoid arthritis in the Japanese population.[J] .Ann. Rheum. Dis., 2020, 79: 103-111.
研究使用较高深度的宏基因组shotgun测序(每个样本平均13 Gb)对日本人群(病例 = 82,对照 = 42)进行了RA肠道微生物组的MWAS分析。MWAS由三个主要的生物信息学分析渠道(系统发育分析、功能基因分析、途径分析)组成。
使用了之前研究中6139个完成拼接日本人肠道宏基因组作为参考序列以及其他几项研究的参考基因组,在过滤部分种过多的基因组之后,最后一共使用了7881个参考基因组。
将QC后的序列直接比对到参考基因组,并根据基因组长度计算对应物种的相对丰度。
基因方面选择denovo组装,使用MegaHIT,然后再contig上完成ORF预测和CD-HIT聚类去冗余,最后与UniRef和KEGG数据库比对。
最后使用bowtie2将测序序列比对到注释后的unigene序列上获得基因丰度,经过KEGG注释得到代谢途径的丰度。研究的数据处理流程图如下:
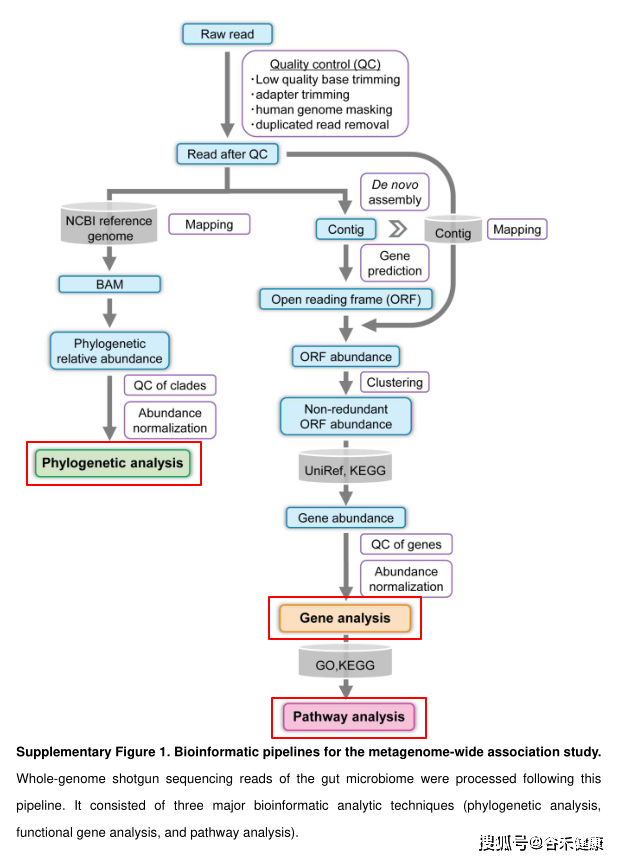
在数据分析流程和方案选择上人体肠道菌群由于研究众多,以及有多个深度测序拼接完成的Binning参考基因组数据集,确实可以直接使用参考基因组直接比对。
对于其他一些环境或来源的样本这个深度的数据量可以考虑独立拼接和分箱。该研究中使用已有参考基因组,大概88%的序列能比对到参考基因组,如果直接组装这个比例应该可以更高一些。另外在获得基因丰度是可以考虑使用Salmon,比对获得基因丰度更为方便。
获得相应数据后对相对丰度,该研究使用Box-Cox transformation对数据进行标准化,并过滤了一些低丰度的菌属。Case-control的相关性分析使用的R的glm2模块,将年龄、性别和测序上机分组作为协变量。
对于菌属的关联分析,最终将显著性结果以火山图和GraPhlAn图的形式展现如下:
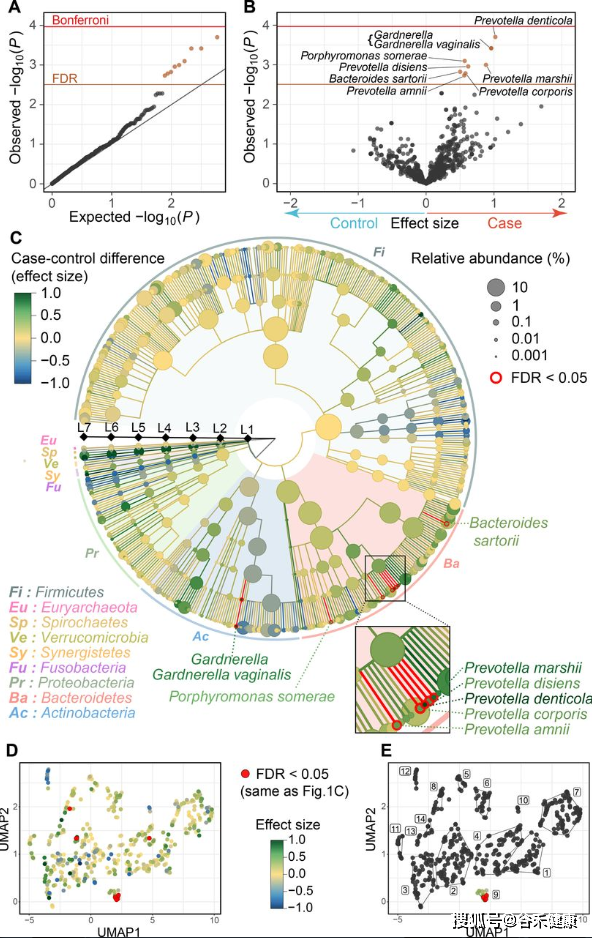
上面其中D图使用每个菌的丰度进行UMAP分析,并映射关联效应的展示比较有意思。
不过在基因层面上并未找到相应的关联,可以看到下图UniRef90的NMDS分布图两组之间无法有效区分,多样性也没有显著差异。
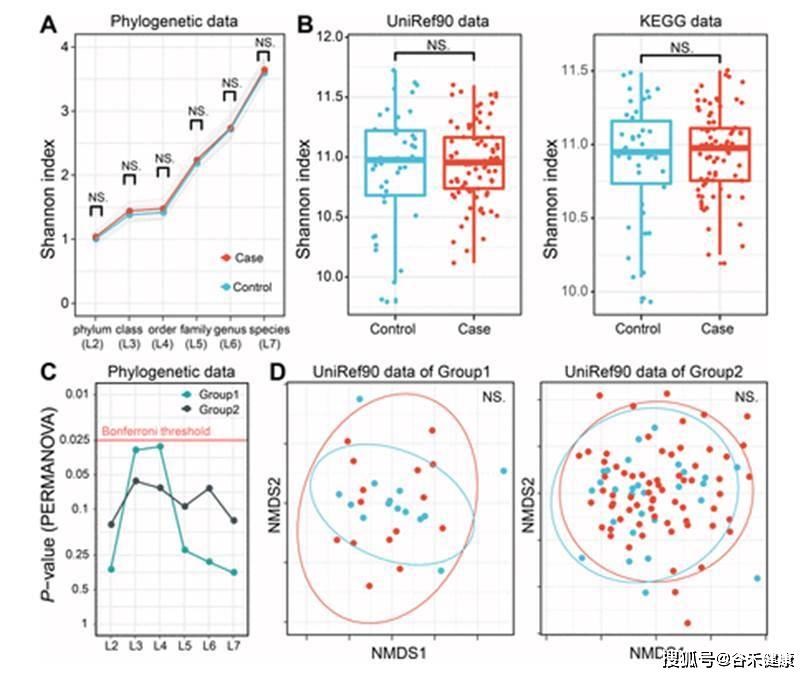
点 评
这项研究在菌层面发现了多个普雷沃氏菌属的菌在日本人群中与类风湿性关节炎存在关联,不过除此之外其他方面的发现并不多,仅找到一个基因存在显著关联,涉及的临床调查也相对有限,且人群队列数量不算多,并无独立验证集,因此亮点并不多。如果能纳入免疫相应指标可能能研究的更细致一些。
< 案例四 > 永冻土中参与有机物降级的关键菌群
最后这项研究是对来自永冻土融化梯度的214个样品的宏基因组测序组装了1,529个基因组,揭示了参与有机物降解的关键种群,包括其基因组编码先前未描述的木糖降解真菌途径的细菌。
文献来源:Woodcroft Ben J,Singleton Caitlin M,Boyd Joel A et al. Genome-centric view of carbon processing in thawing permafrost.[J] .Nature, 2018, 560: 49-54.
通过宏基因组denovo组装和分箱Binning,最终获得了1529个永冻土菌群基因组。基于这些数据描绘了永冻土融化梯度下的菌群构成特征,如下图。
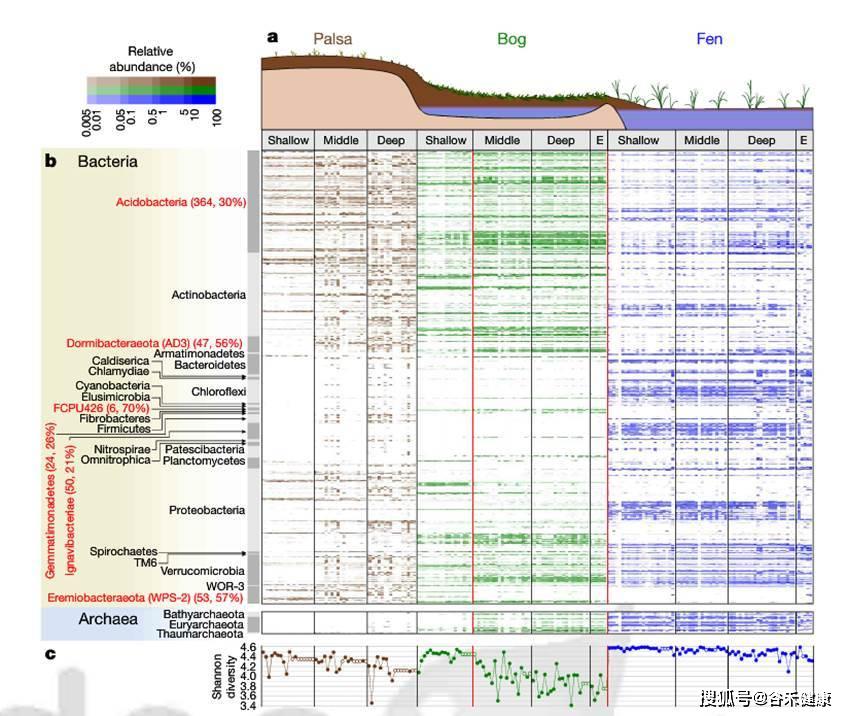
论文是2018年发表的,测序是在2011和12年测的,使用的是CLC Genomics Workbench 较早的4.4版分单样本组装,然后使用MetaBAT进行分箱,最后的标准是70%完成度和低于10%的污染。
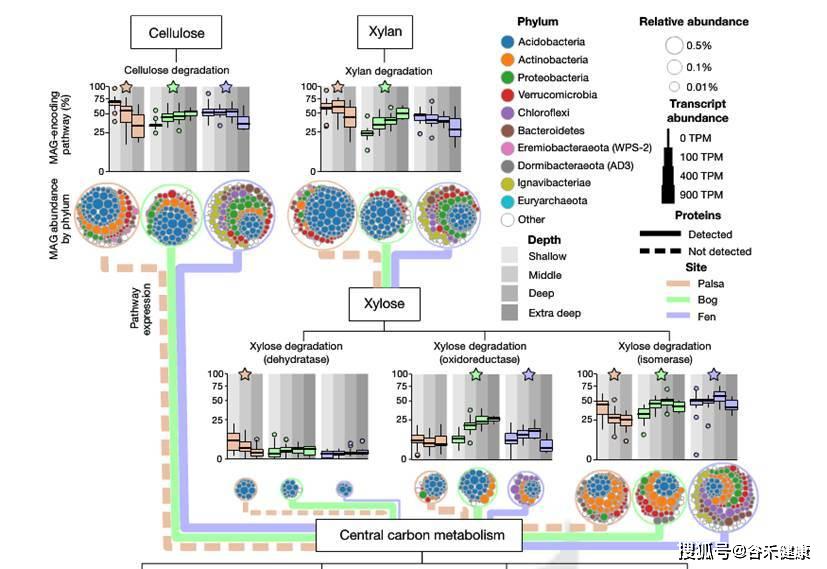
其中糖苷水解酶基因使用dbCAN数据库的HMM进行预测,碳代谢使用KEGG数据。
研究还同时选择了部分样本进行了宏转录组和宏蛋白组的测序,对碳代谢同时结合转录组和蛋白组的数据,展现了特定通路下不同永冻土的菌群构成和表达丰度差异。
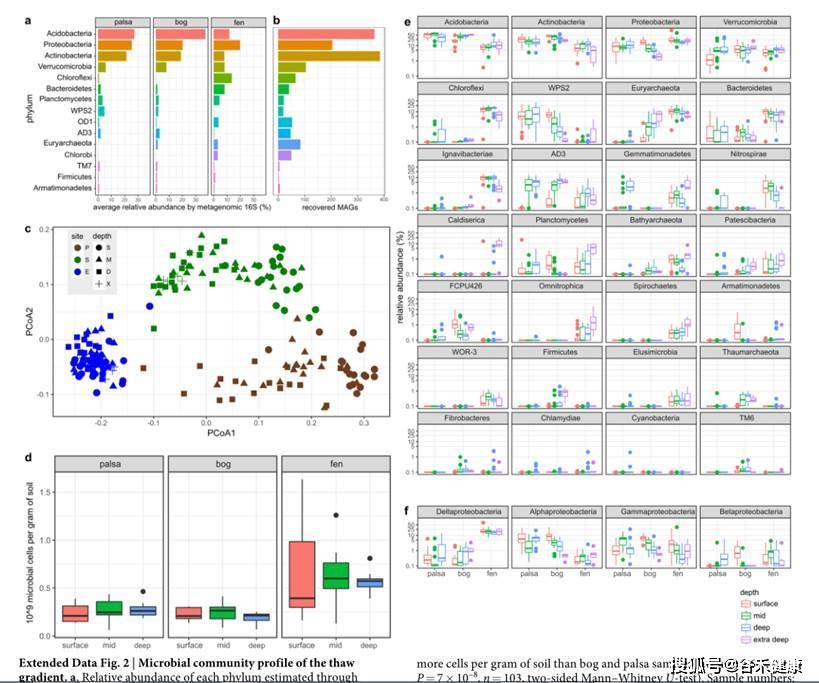
基因组拼接的分布情况,以及不同地域样本分布和菌属丰度情况如下:
木糖降解途径在每个样本中的分布和维恩图,另外详细的展现了主要门对每个代谢途径的贡献和基因表达丰度,如下图:
这张图分析了特定菌与地理位置和CO2以及甲烷的浓度的关联性,如下图:
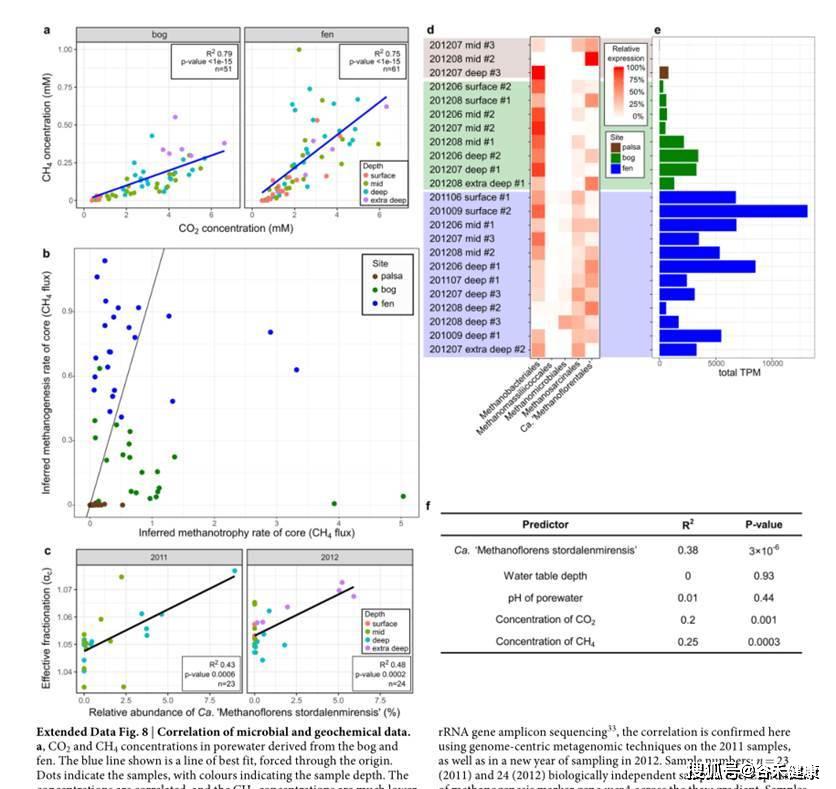
对关键物种的CH 4 :CO2浓度比相关代谢途径的重建,以及相应基因的基因家族分析。
点 评
总结一下这项研究,永冻土的菌群参考基因组数据缺乏,该研究从大量地点采集样本重建了1500多个参考基因组。
首先从物种构成特征上与永冻土融化阶段特征进行分析,并与重要环境因子进行分析,锁定重要的特征菌。
然后针对重要的代谢途径和关键基因结合宏转录组和宏蛋白组全面解析代谢途径的分布和差异变化。对关键的物种重建了相关代谢途径并对其相关基因家族进行分析。
研究基本上从头构建了一个生态环境下的菌群结构数据,并利用获得的基因组深度解析特定代谢途径和基因的构成和表达变化,应该说既宽又深。很多样本采集和测序是2011年和 2012年就开展的,虽然测序技术远不如现在成本低和成熟,但是其独特的研究对象和全面深入的分析仍然使整项研究和目前的一些研究相比完成的更加出色。
p.s. 以上展示的图表,我们都可以帮你实现~
05
工具分享
一、 MicrobiomeAnalyst
网址:https://www.microbiomeanalyst.ca/,只需要biom文件或丰度表就可以进行绝大部分统计检验分析,而且生成图表完整,可以直接使用。偶尔会有服务器不稳定,上传提示错误的情况。
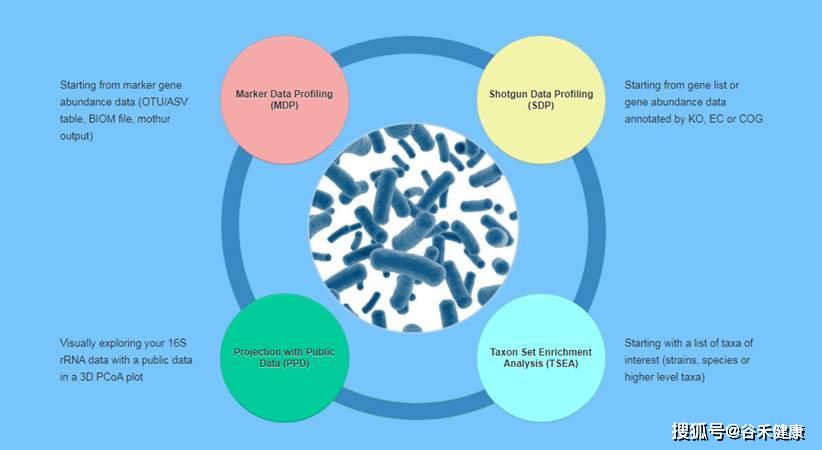
特别推荐其中的Taxon Set Enrichment Analysis模块,直接提交物种列表(一般是找到的差异物种列表),可以直接在各种已有的相关性(人体基因-菌属相关,生活方式-菌属相关,疾病-菌属相关)中进行富集分析,能很好的帮助判断和提供差异菌群的具体关联和证据支持。
完整的支持分析包括:

可以直接生成下面的图:
基本上常见的分析和图都能在线实现。
二、 gcMeta
另一个是https://gcmeta.wdcm.org/,是中科院微生物研究所搞的平台,里面包括了宏基因组的样本数据和在线分析平台,可以直接上传原始数据,直接使用工具进行在线分析,大部分常见工具都有,也有一些流程。
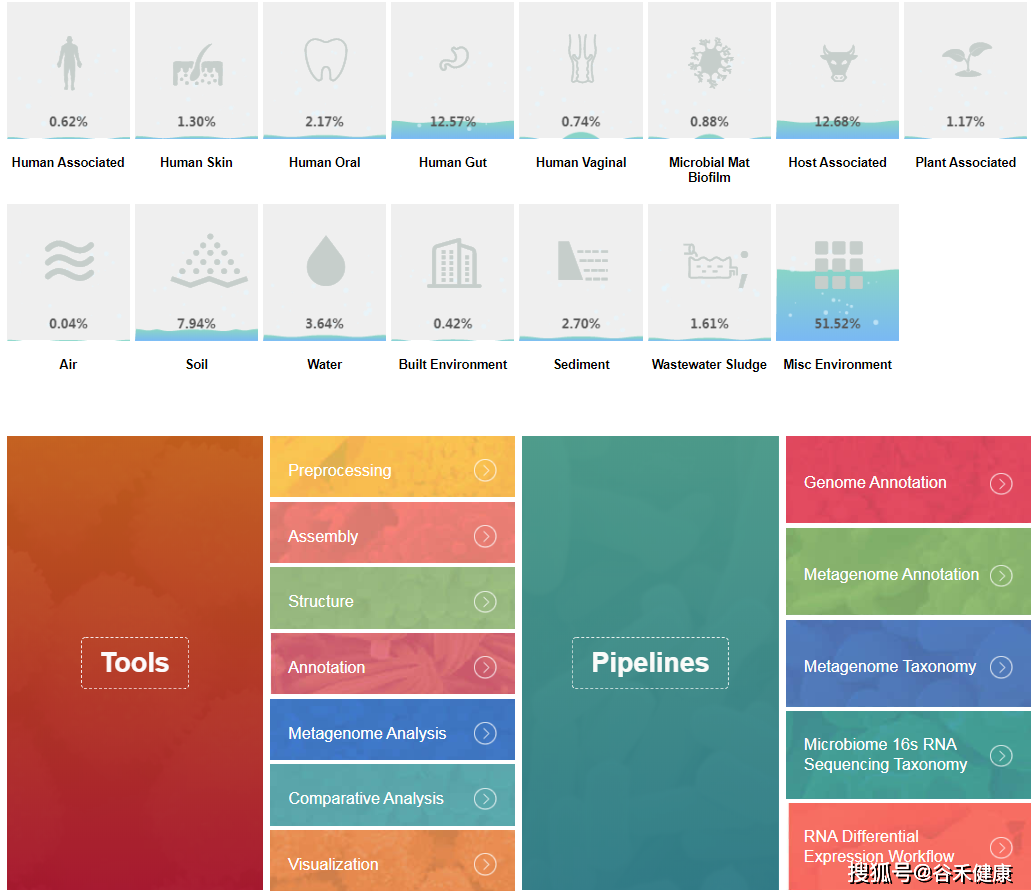
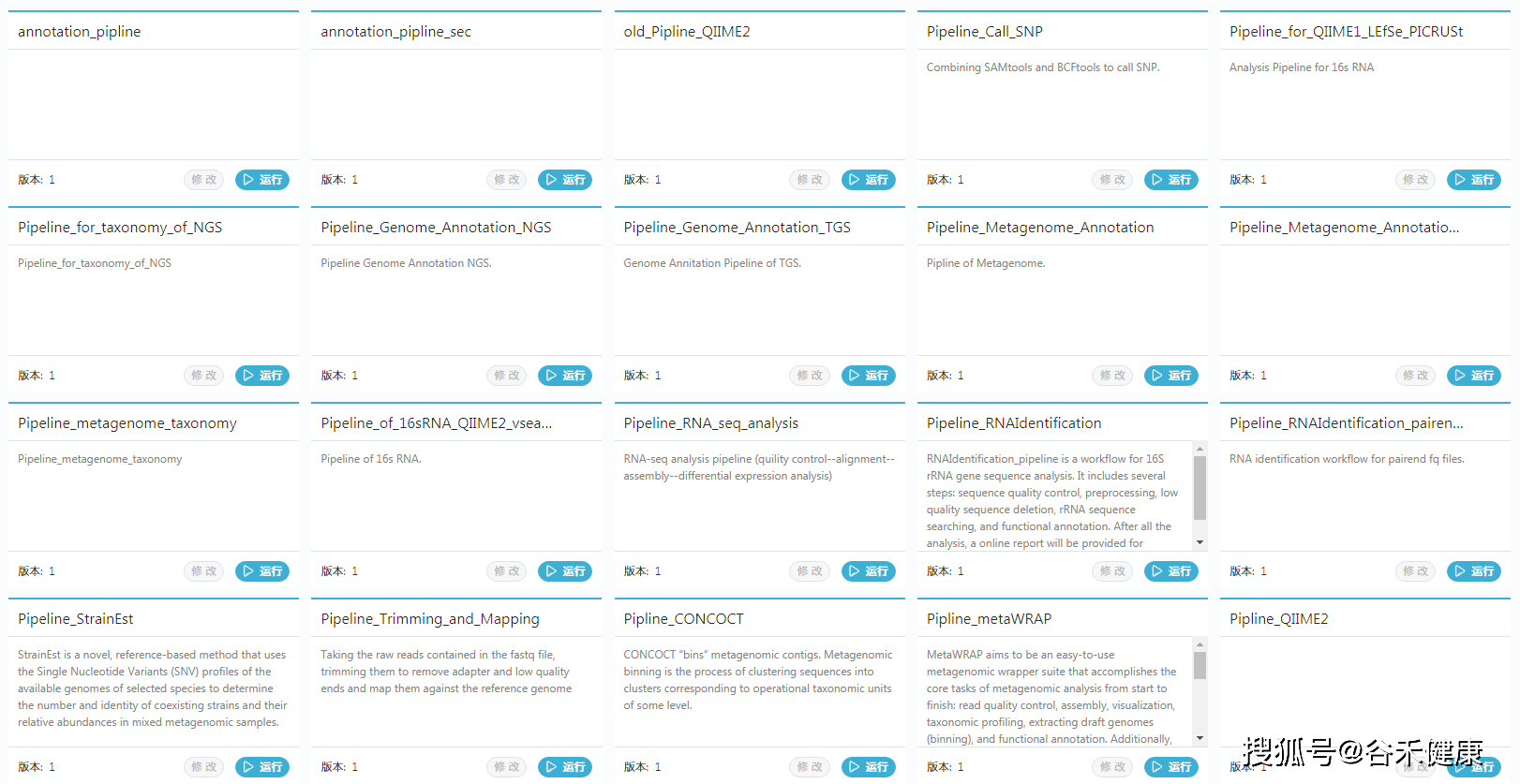
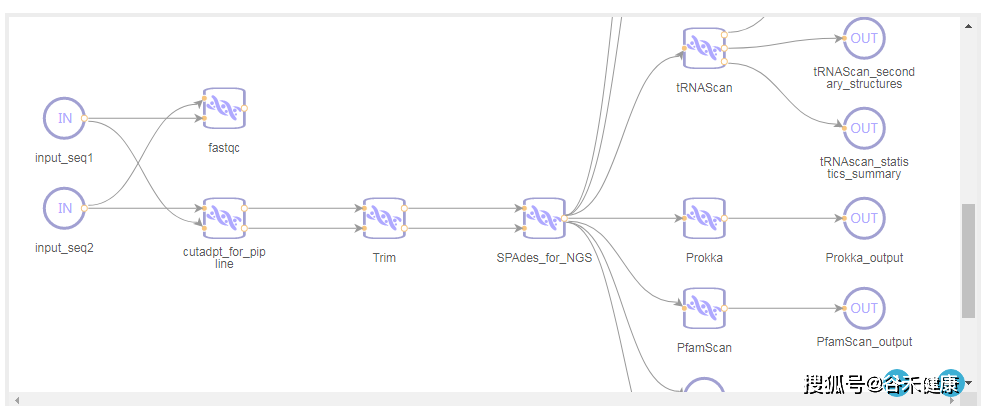
对于缺乏计算资源或想自己动手分析的朋友挺友好的,非常推荐试试看。
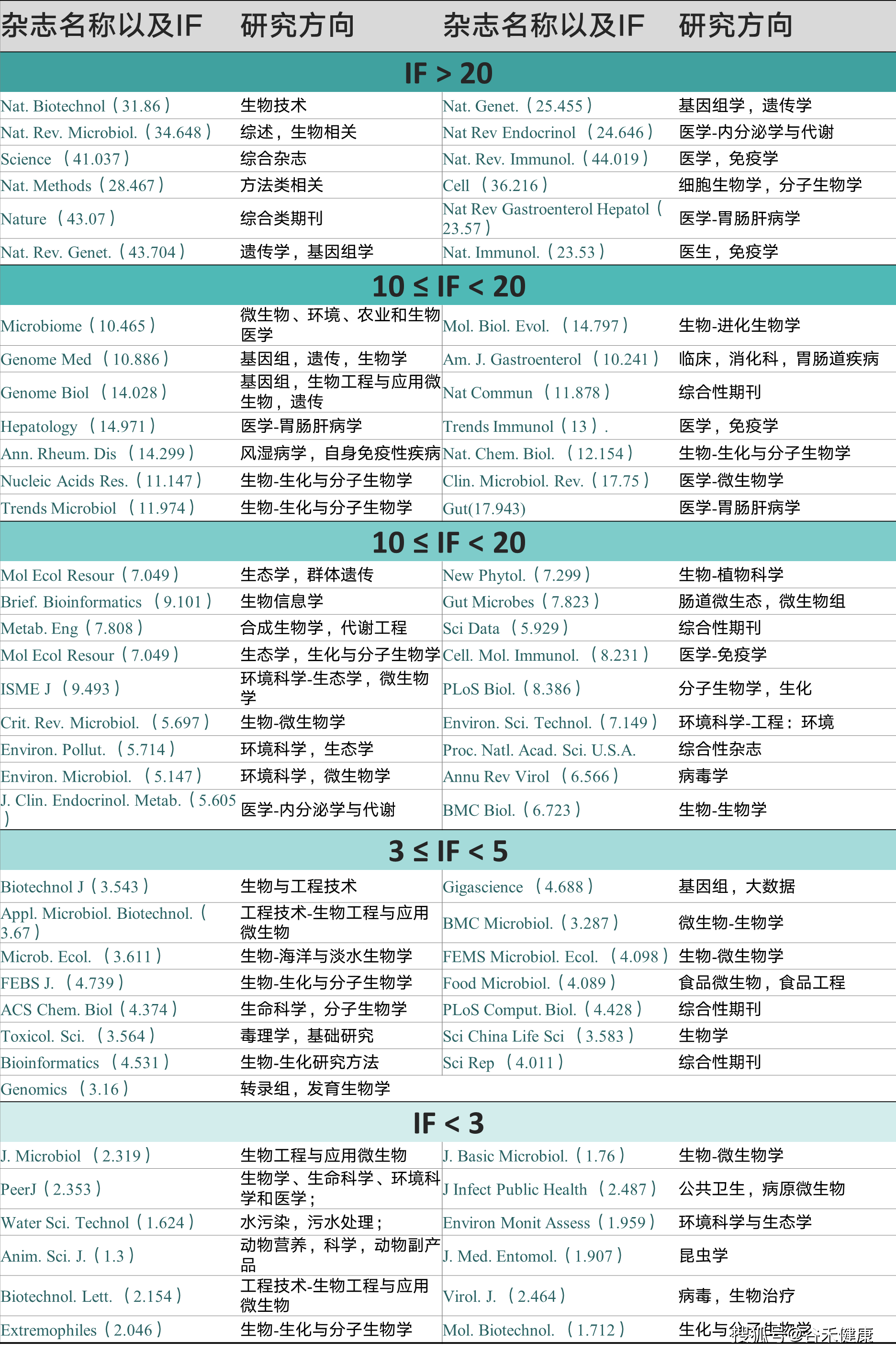
最后,帮大家整理了宏基因组可投稿的期刊,具体研究方向和影响因子见下表:

- 本文固定链接: https://maimengkong.com/zu/753.html
- 转载请注明: : 萌小白 2021年8月20日 于 卖萌控的博客 发表
- 百度已收录
