组装是宏基因组分析中最重要的一环,它直接影响着后续基因集的构建、功能分析及物种注释等的分析结果。
小锐使用一个大约12G的土壤样本,测试了SOAPdenovo、megahit和metaSAPdes三个组装软件的组装性能。到底谁能胜出?请随着小锐一步一步来探个究竟。
1
软件安装
SOAPdenovo下载地址
http://sourceforge.net/projects/soapdenovo2/files/SOAPdenovo2/
MEGAHIT下载地址
https://github.com/voutcn/megahit
metaSPAdes下载地址
http://spades.bioinf.spbau.ru/release3.11.0/
评估软件quast下载地址
https://github.com/ablab/quast.git -b release_4.5
2
组装
SOAPdenovo组装
组装命令
SOAPdenovo-63mer all –s sample.config -p 10 -d 1 -K 55 -M 3 -F -u –o sample
参数说明
-s config配置文件
-p 需要使用的cpu数目,默认8
-K k-mer的长度(选取了4个kmer,分别是51,55.59,63)
-d [INT], kmerFreqCutoff, 去除频数小于等于该值的kmers,默认为0
-M [INT], mergeLevel连接contigs时, 合并相似序列的等级,默认为1,最小值为0,最大值为3
-F 利用reads对scaffolds的gap进行填补,默认不执行
-u 构建scaffold前屏蔽过高或过低覆盖度contigs,默认屏蔽
-o 输出文件前缀
MEGAHIT 组装
组装命令
megahit [options] {-1 <pe1> -2 <pe2> | --12 <pe12> | -r <se>} [-o <out_dir>]
参数说明
[options] 可以根据宏基因组数据的类型选择不同的类型,-1代表reads1,-2 代表reads2,-r 是single reads ; -o 是输出文件夹。
其他参数默认就可以,k-mer的长度是[21,29,39,59,79,99,119,141]。
metaSPAdes组装
组装命令
spades.py --meta -1 sample.1.fq.gz -2 sample.2.fq.gz -s sample.single.fq.gz -t -m -k -o <dirname>
参数说明
--meta 是宏基因组的组装(该程序可以进行其他类型数据的应用,比如RNA),-1,-2,-s 输入的序列,-t 使用的cpu数目(默认16),-m 使用的最大内存限制(默认250G),-k k-mer的长度(默认自动增加,最大128),-o 输出文件夹。
3
评估组装结果
接下来,就到大家最关心的时刻了。
马上奉上测评结果!

组装使用的时间及计算资源
3款软件对比
组装软件
时间
内存
SOAPdenovo
约24h
220G
metaSAPdes
约48h
250G
megahit
约6h
30G
组装性能
下载评估软件quast,解压即可进行评估。
运行QUEST
quast.py sample.contigs.fa -o report
输入组装得到的结果.contigs.fna文件(我们选取了contigs大于500bp的序列),-o 输出文件的位置。
各个组装软件具体结果评估并比较如下:
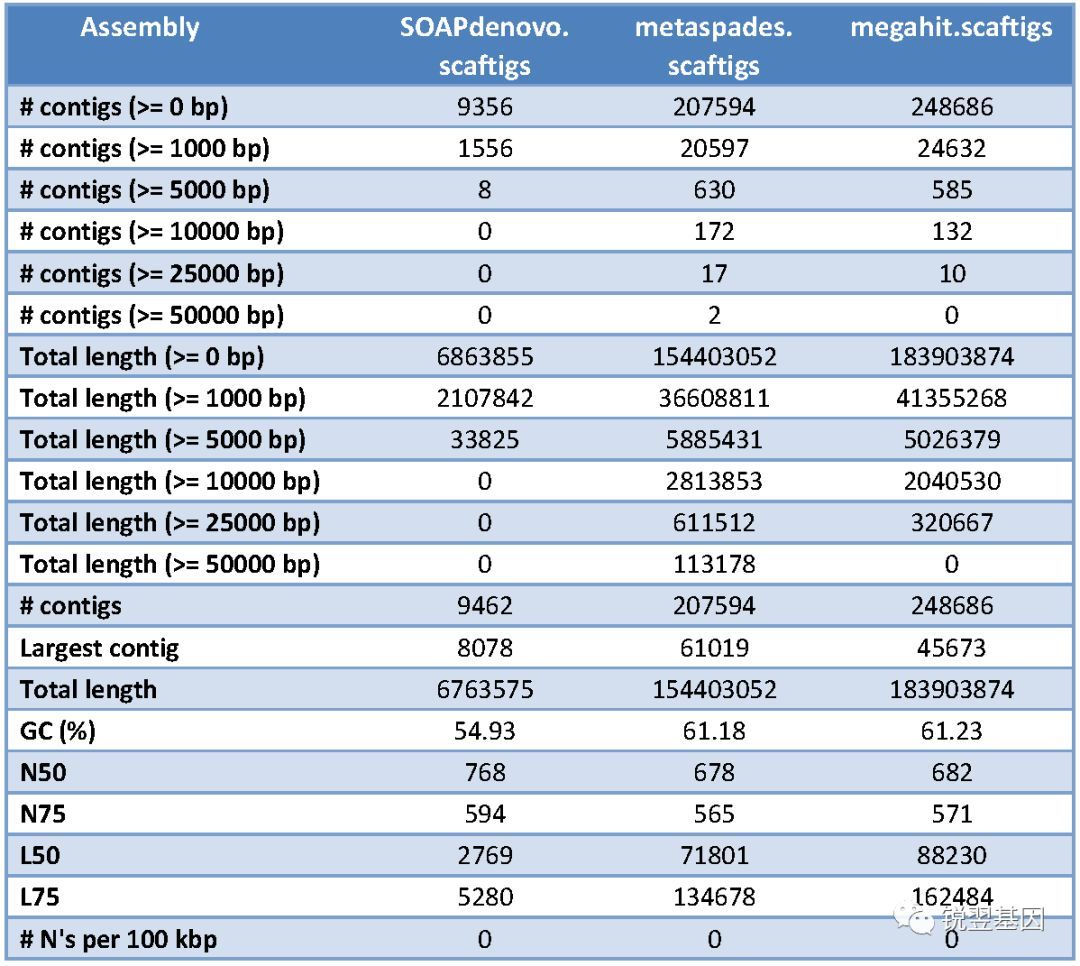
- 总结 -
小锐教你怎么选
从组装性能上看,三个不同的组装软件具有不同的特点:
SOAPdenovo组装序列的contigs数相对较少,适用一些简单样本的组装(比如人的肠道样本)。
而metaSAPdes和megahit组装得到的contigs数较多,适用一些复杂样本的组装(比如土壤和水体等样本)。
但同时考虑到组装过程中的计算时间及资源,megahit软件相对具有更好的组装性能。
- 本文固定链接: https://maimengkong.com/zu/752.html
- 转载请注明: : 萌小白 2021年8月19日 于 卖萌控的博客 发表
- 百度已收录
