科研数据分析中,如何知道你的差异基因或得到的基因列表富集在什么组织中?今天分享的TissueEnrich包可以轻松帮你解答这个问题。我们一起来学习一下这个R包。
TissueEnrich包简介
TissueEnrich包用于计算一组输入基因中的组织特异性基因富集程度。例如,用户可以输入RNA-Seq数据中表达量最高的基因或基因共表达模块,以确定在这些数据集中的基因富集到了哪些组织特异性。组织特异性基因的定义来源于对人类蛋白质图谱(HPA)(Uhlén等,2015)、GTEx(Ardlie等,2015)和小鼠ENCODE(Shen等,2012)的RNA-Seq数据进行处理,并采用了HPA的算法(Uhlén等,2015)。超几何检验用于确定输入基因中是否富集了组织特异性基因。除了组织特异性基因的富集分析,TissueEnrich包还可以使用用户提供的表达数据集来定义组织特异性基因,并进一步计算组织特异性基因富集。TissueEnrich包含以下三个功能:
-
teEnrichment:给定一个基因列表作为输入,该功能使用来自人类或小鼠RNA-Seq数据集的组织特异性基因来计算组织特异性基因的富集程度。
teEnrichment函数用于计算输入基因集中的组织特异性基因富集情况。使用通过处理人类和小鼠的RNA-Seq数据集定义的组织特异性基因。用户必须在输入的GeneSet对象中通过
organism参数指定物种(“Homo Sapiens”(默认)或“Mus Musculus”)。在使用teEnrichment时,用户可以指定用于组织特异性基因富集分析的RNA-Seq数据集(
rnaSeqDataset):-
1 表示“Human Protein Atlas”(默认) -
2 表示“GTEx” -
3 表示“Mouse ENCODE”
数据类型: 使用人类或小鼠的组织特异性基因(如HPA、GTEx、或小鼠ENCODE数据集)进行富集分析。
输入: 输入基因集(可以是基因符号或Ensembl ID),以及用户指定的RNA-Seq数据集(例如,HPA、GTEx或小鼠ENCODE)。
输出: 计算并返回富集结果,显示每种组织中特异性基因的富集情况。
-
-
teGeneRetrieval:给定跨组织的基因表达数据,该功能使用HPA的算法来定义组织特异性基因。
数据类型: 使用来自HPA(或其他指定的RNA-Seq数据集)定义的组织特异性基因。
输入: 用户提供的基因表达数据(如SummarizedExperiment对象),数据可以包含多个组织的表达信息。
输出: 返回包含每个基因在不同组织中的组织特异性信息的结果,显示基因所属的组织特异性组(如“组织富集”、“组富集”或“组织增强”等)。
-
teEnrichmentCustom:给定基因列表和来自teGeneRetrieval定义的组织特异性基因作为输入,该功能计算组织特异性基因的富集。
数据类型: 使用teGeneRetrieval函数返回的组织特异性基因,结合用户提供的自定义基因表达数据进行富集分析。
输入: 自定义的输入基因集(如来自文件的基因列表)和由teGeneRetrieval生成的组织特异性基因信息。
输出: 返回一个包含富集分析结果的列表对象,显示组织特异性基因的富集情况,并可进一步使用ggplot等工具进行可视化。
组织特异性基因的定义
组织特异性基因根据HPA的算法(Uhlén等,2015)定义,并可分为以下几类:
-
组织富集基因:表达水平大于1(TPM或FPKM),并且在特定组织中的表达水平至少比其他所有组织高5倍。 -
组富集基因:表达水平大于1(TPM或FPKM),在2-7个组织中的表达水平至少比其他所有组织高5倍,但不被视为组织富集基因。 -
组织增强基因:表达水平大于1(TPM或FPKM),在特定组织中的表达水平至少比所有其他组织的平均水平高5倍,但不被视为组织富集基因或组富集基因。
在teEnrichment中,用户可以指定用于组织特异性基因富集分析的基因类型(tissueSpecificGeneType):
-
1 表示“All” (default) -
2 表示 “Tissue-Enriched” -
3 表示“Tissue-Enhanced” -
4 表示 “Group-Enriched”
本节内容,我们主要使用第一种功能进行组织特异性基因富集分析,首先一起来安装TissueEnrich包。
TissueEnrich包安装
方法一:直接从BIOC下载安装(推荐)
BiocManager::install("TissueEnrich") 方法二:从Github下载安装
#Install Dependencies install.packages(c("dplyr","ensurer","ggplot2","tidyr"))
install.packages("BiocManager")
BiocManager::install("SummarizedExperiment")
BiocManager::install("GSEABase") #Now install the devtools package install.packages("devtools") library(devtools)
install_github("Tuteja-Lab/TissueEnrich") 
安装成功可使用 vignette("TissueEnrich") 命令查看帮助文档。
TissueEnrich 包的使用
这里我们以R包提供的数据为例,使用一组包含100个基因的列表进行组织特异性富集分析,探讨这些基因富集在哪类组织中。
人类胚胎前期发育单细胞数据
数据为TissueEnrich R保自带,使用了来自人类胚胎前期发育(在受精后第5、6和7天)的单细胞RNA-Seq分析中鉴定出的滋养层(Trophectoderm, TE)特异性基因(Petropoulos等,2016)。通过主成分分析(PCA)将单细胞分配到内细胞团(包括外胚层和新兴的外胚内胚层)和滋养层中。接着,通过差异基因表达分析,生成了100个TE特异性基因的列表(Petropoulos等,2016)。我们将这100个基因作为输入基因集,使用HPA数据集定义的组织特异性基因进行组织特异性基因富集分析。
数据集说明
-
输入基因集可以包含Ensembl Ids(ENSEMBLIdentifier())或基因符号(SymbolIdentifier()),在GeneSet对象中通过 geneIdType参数来指定这一点。
library(TissueEnrich) # 读取输入基因列表 genes<-system.file("extdata",
"inputGenes.txt",
package = "TissueEnrich")
inputGenes<-scan(genes,character()) # 创建基因集对象 gs<-GeneSet(geneIds=inputGenes,
organism="Homo Sapiens",
geneIdType=SymbolIdentifier()) # 执行TE富集分析 output <- teEnrichment(inputGenes = gs) 到这里,我们就计算完了TE的富集结果,得到了一个list对象。
组织特异性基因富集结果分析
teEnrichment函数的输出是一个包含富集结果的列表对象,接下来,我们可以使用这些结果进行可视化。
使用ggplot2绘制组织特异性基因富集条形图
富集结果的第一个对象是一个SummarizedExperiment对象,其中包含对应于组织特异性基因富集的−Log10(P−Value)和fold-change值,以及输入基因集中的组织特异性基因的数量。
#提取组织特异性基因分析结果 seEnrichmentOutput<-output[[1]]
enrichmentOutput<-setNames(data.frame(assay(seEnrichmentOutput),row.names = rowData(seEnrichmentOutput)[,1]), colData(seEnrichmentOutput)[,1])
enrichmentOutput$Tissue <- row.names(enrichmentOutput) 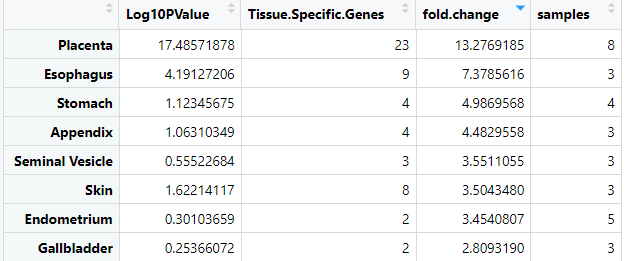
可以从表格中看出,我们提供的基因列表主要集中在Placenta 胎盘组织中,这与我们的数据类型相一致,接下来我们可以使用组织特异性富集分析结果通过ggplot2绘制组织特异性基因富集的条形图。
# 绘制−Log10(P−Value)的条形图 ggplot(enrichmentOutput, aes(x = reorder(Tissue, -Log10PValue), y = Log10PValue, label = Tissue.Specific.Genes, fill = Tissue)) +
geom_bar(stat = 'identity') +
labs(x = '', y = '-LOG10(P-Adjusted)') +
theme_bw() +
theme(legend.position = "none") +
theme(plot.title = element_text(hjust = 0.5, size = 20), axis.title = element_text(size = 15)) +
theme(axis.text.x = element_text(angle = 45, vjust = 1, hjust = 1), panel.grid.major = element_blank(), panel.grid.minor = element_blank()) 
结果解释
图中x轴表示每个组织,y轴表示组织特异性基因富集的−Log10(P−Value)值。可以看到,100个TE特异性基因在胎盘特异性基因上显示了显著的富集。
同样,我们可以使用富集的fold-change值绘制条形图,来进一步展示不同组织间的变化情况。
ggplot(enrichmentOutput,aes(x=reorder(Tissue,-fold.change),y=fold.change,label = Tissue.Specific.Genes,fill = Tissue))+
geom_bar(stat = 'identity')+
labs(x='', y = 'Fold change')+
theme_bw()+
theme(legend.position="none")+
theme(plot.title = element_text(hjust = 0.5,size = 20),axis.title = element_text(size=15))+
theme(axis.text.x = element_text(angle = 45, vjust = 1, hjust = 1),panel.grid.major= element_blank(),panel.grid.minor = element_blank()) 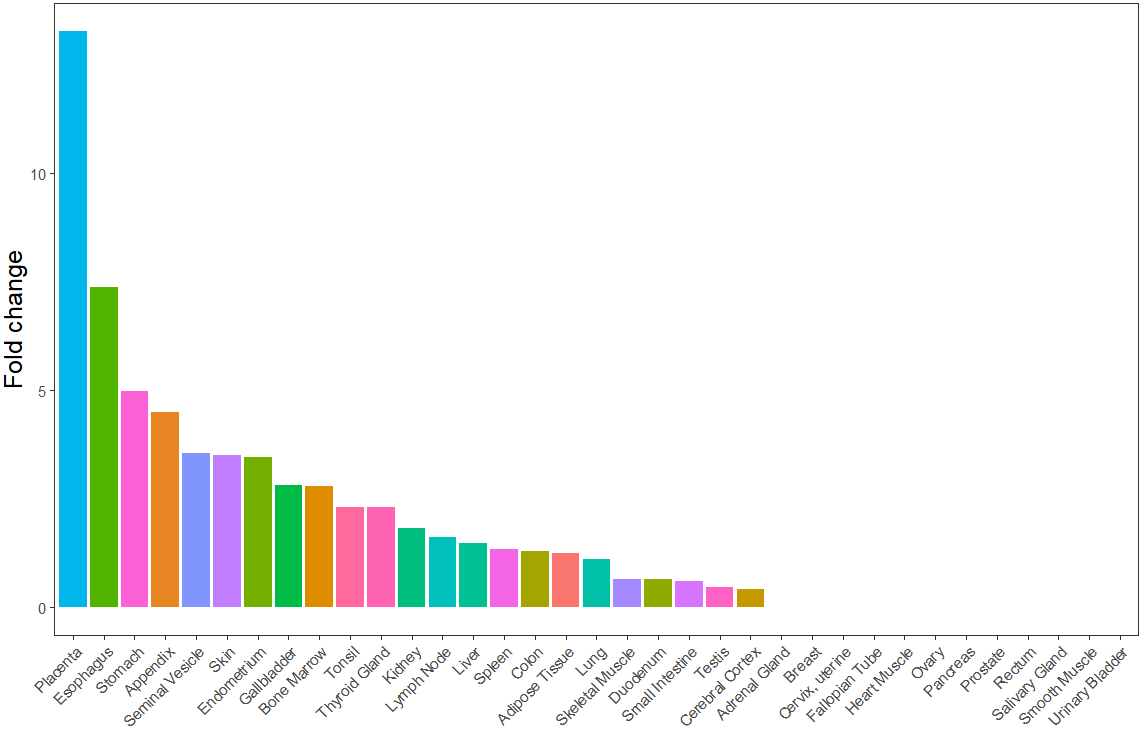
Fold change图显示了组织特异性富集的Fold change差异倍数。
组织特异性基因的热图展示
富集结果的第二个对象包含了输入基因集中组织特异性基因的表达值。我们可以将这些表达值可视化为热图,进一步展示不同组织中基因的表达情况。
library(tidyr) # 获取胎盘组织的表达数据 seExp <- output[[2]][["Placenta"]]
exp <- setNames(data.frame(assay(seExp), row.names = rowData(seExp)[, 1]), colData(seExp)[, 1])
exp$Gene <- row.names(exp)
exp <- exp %>% gather(key = "Tissue", value = "expression", 1:(ncol(exp) - 1)) # 绘制热图 ggplot(exp, aes(Tissue, Gene)) +
geom_tile(aes(fill = expression), colour = "white") +
scale_fill_gradient(low = "white", high = "steelblue") +
labs(x = '', y = '') +
theme_bw() +
guides(fill = guide_legend(title = "Log2(TPM)")) +
theme(plot.title = element_text(hjust = 0.5, size = 20), axis.title = element_text(size = 15)) +
theme(axis.text.x = element_text(angle = 45, vjust = 1, hjust = 1), panel.grid.major = element_blank(), panel.grid.minor = element_blank()) 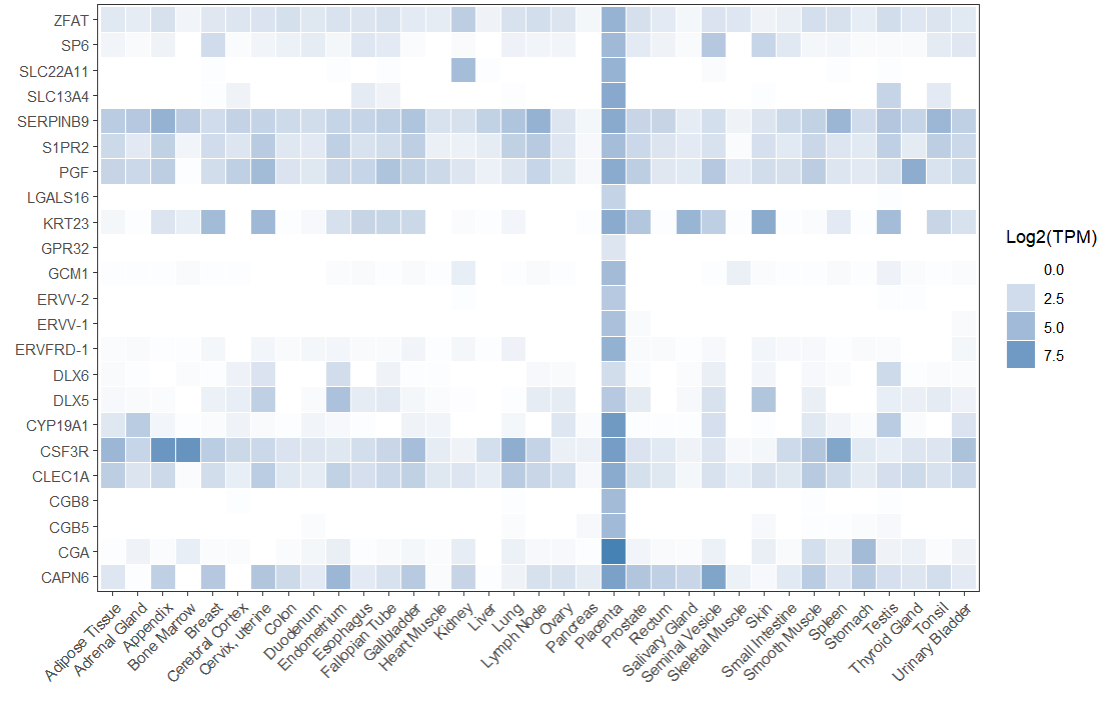
提取输入基因的组织特异性信息
如何提取富集到Placenta的基因呢?
输出的第三个对象是一个列表,包含了输入基因的组织特异性信息。以下代码可以检索输入基因在Placenta组织中的特异性信息。
# 获取胎盘组织的组织特异性基因信息 seGroupInf <- output[[3]][["Placenta"]]
groupInf <- data.frame(assay(seGroupInf))
print(head(groupInf)) #> Gene Group #> 1 CGA Tissue-Enriched #> 2 GCM1 Tissue-Enriched #> 3 CYP19A1 Tissue-Enriched #> 4 GPR32 Tissue-Enriched #> 5 CLEC1A Tissue-Enriched #> 6 SLC13A4 Tissue-Enriched 匹配到其他组织的基因提取方法同上。
提取无法映射到组织特异性基因数据的基因
输出列表的第四个对象是一个字符向量,包含无法在组织特异性基因数据中找到的输入基因。以下代码检索这些基因。
# 检索无法映射到组织特异性基因数据的基因 print(geneIds(output[[4]])) #> [1] "C10ORF54" "CGB" "GRAMD3" "PVRL4" 以上是进行人类基因列表在人类组织特异性基因表达的分析。
人的基因能否通过基因名和小鼠的组织特异性基因进行富集分析呢?
以下是案例:
使用人类基因列表进行小鼠组织特异性基因富集分析
在本节,我们展示了如何使用 TissueEnrich 包通过输入的人类基因数据,进行小鼠组织特异性基因富集分析。具体来说,我们使用了之前示例中的100个胚胎外层(TE)特异性基因列表,来探索使用小鼠ENCODE数据进行的组织特异性基因富集分析。
步骤
1.加载R包 首先,我们加载必要的R包,包括用于组织特异性基因富集分析的 TissueEnrich 包和用于可视化结果的 ggplot2包。
library(TissueEnrich)
library(ggplot2)
2.输入基因数据 输入的基因列表是一个包含基因符号的文本文件。在这个例子中,我们使用的是从人类胚胎着床数据集中(Petropoulos et al. 2016)识别的100个TE特异性基因。我们使用 scan 函数将基因数据加载到R中。
genes <- system.file("extdata", "inputGenes.txt", package = "TissueEnrich")
inputGenes <- scan(genes, character())
3.创建GeneSet对象 我们创建一个 GeneSet 对象,这个对象是输入基因列表的容器,指定了物种(Homo sapiens)和基因标识符类型(SymbolIdentifier,使用基因符号)。
gs <- GeneSet(geneIds = inputGenes,
organism = "Homo Sapiens",
geneIdType = SymbolIdentifier())
4.进行组织特异性基因富集分析 接下来,我们使用 teEnrichment 函数进行组织特异性基因富集分析,提供基因集作为输入,并指定RNA-Seq数据集为小鼠(数据集3,对应ENCODE数据)。该函数返回包含富集结果的列表,包括富集统计信息和倍数变化值。
output <- teEnrichment(inputGenes = gs, rnaSeqDataset = 3)
5.提取并可视化结果 输出列表中的第一个对象是一个 SummarizedExperiment 对象,包含每个组织的−Log10(P-value)和倍数变化值。我们提取这些值,并使用 ggplot2 创建一个条形图,展示组织特异性基因富集情况。
#提取结果 seEnrichmentOutput <- output[[1]]
enrichmentOutput <- setNames(data.frame(assay(seEnrichmentOutput), row.names = rowData(seEnrichmentOutput)[, 1]), colData(seEnrichmentOutput)[, 1])
enrichmentOutput$Tissue <- row.names(enrichmentOutput) #可视化显著性结果 ggplot(enrichmentOutput, aes(x = reorder(Tissue, -Log10PValue), y = Log10PValue, label = Tissue.Specific.Genes, fill = Tissue)) +
geom_bar(stat = 'identity') +
labs(x = '', y = '-LOG10(P-Adjusted)') +
theme_bw() +
theme(legend.position = "none") +
theme(plot.title = element_text(hjust = 0.5, size = 20), axis.title = element_text(size = 15)) +
theme(axis.text.x = element_text(angle = 45, vjust = 1, hjust = 1), panel.grid.major = element_blank(), panel.grid.minor = element_blank()) 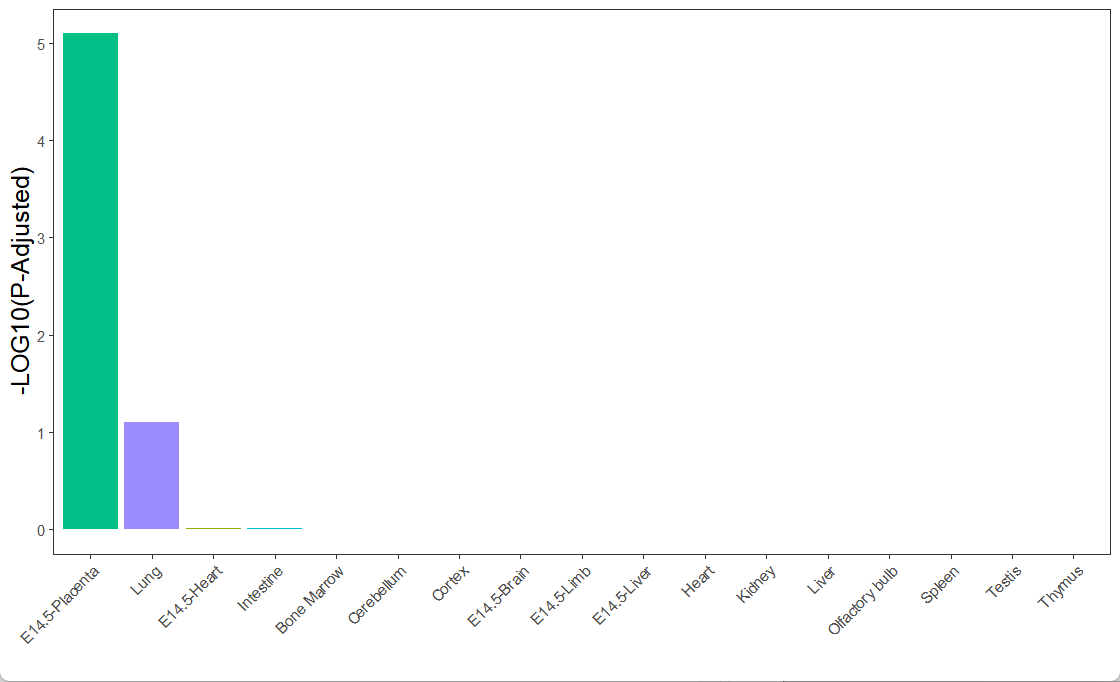
该条形图显示了不同小鼠组织中的组织特异性基因富集情况,其中人类TE特异性基因在小鼠胎盘特异性基因中表现出富集。y轴表示−Log10(P-value),指示富集的统计显著性,x轴显示不同的组织。
结果解释
从结果图中可以清晰地看到,人类TE特异性基因在小鼠胎盘组织中表现出富集,反映了人类和小鼠之间组织特异性基因表达的高度保守性。这表明,人类TE特异性基因列表可以作为探索小鼠组织中类似基因表达模式的参考。(仅建议参考)
以上是如何使用TissueEnrich包进行基因集的组织特异性表达富集分析的全部介绍,TissueEnrich包还有计算基因在组织、组中富集的方法,我们将在下一期介绍。
Reference: Jain, A, Tuteja, G. (2018) TissueEnrich: Tissue-specific gene enrichment analysis. Bioinformatics, bty890. 10.1093/bioinformatics/bty890
- 本文固定链接: https://maimengkong.com/zu/1826.html
- 转载请注明: : 萌小白 2024年10月25日 于 卖萌控的博客 发表
- 百度已收录
