一、WGCNA分析简介
随着转录组测序价格下降,进行测序的样本数也在逐渐增多,WGCNA(weighted gene co-expression networkanalysis,权重基因共表达网络分析)这种适合大样本的分析方法,在疾病以及其他性状与基因关联分析等方面应用越来越广泛。
WGCNA最大的优势在于,能将多个样本中成千上万的基因,根据它们的表达模式划分到数个至数十个模块当中,然后以模块为单位进行分析,降低了我们的运算量,也提高了准确性。
每个模块基因都有特定的表达模式,我们对获得的模块可以进行两两之间或是样品与模块之间相关性和聚类分析,了解模块的具体情况。若有性状等表型信息,还可以分析模块与性状间的关系,找出与性状最相关的模块。
我们做WGCNA的分析的时候,还能得到各基因的连通性,可以选择模块中的核心基因或是我们关注的目标基因,结合注释信息,比如分析转录因子(TF)和其他基因之间的调控关系,找到关键的TF,进一步了解TF的调控机制。
如果觉得自己分析起来太复杂又不全面,基迪奥能提供专业的WGCNA分析流程,近期我们对此又做了更新,能给出几乎所有的重要结果。除了上述提到的分析步骤,我们还会对各模块进行GO/KO富集分析。我们可以挑选富集到我们重点关注的GO term或者Pathway的模块,依据基因的功能再进一步分析。
另外一个更重要的分析点,是利用各模块内核心基因连通性绘制网络调控图,将复杂的表格信息可视化,刚才提到的TF的分析,报告中都会给出TF的统计结果,便于在调控网络图绘制时进行标注(图1)。
通常利用WGCNA找到我们重点关注的目标模块和基因,加上一些qPCR等简单的实验验证,就能帮助我们相对轻松地发一篇三分以上的文章。下面我们就以一篇客户文章为例,看看这类性价比较高的文章,是按照怎样的思路完成的。
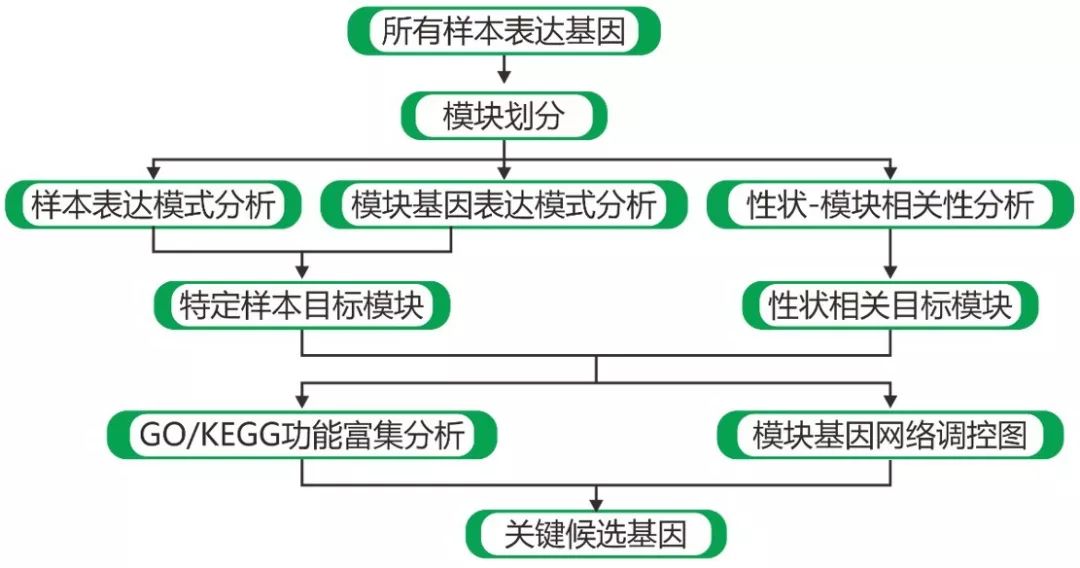
图1 WGCNA总体分析流程图
二、WGCNA案例
虾夷扇贝,Patinopecten(Mizuhopecten)yessoensis是一种重要的经济水产物种,人们发现该物种的雌雄同体可以通过自交系的构建来改善各种性状,但对该物种性别决定和分化机制的了解非常有限,阻碍了这些改进。
作者发现在其他软体动物中(Pacific oyster,banana shrimp,Chinese tongue sole)有应用WGCGNA找到性别偏倚的模块,并通过网络图找到一系列核心基因的案例。所以在这个研究中,作者也应用WGCNA分析扇贝雌雄个体的三个性腺发展阶段基因的表达,找到了对性别分化起到关键作用的基因。
作者对6组共18个样本做了转录组测序,去除掉了分化异常的样本,保留了高质量的序列。然后按照转录组常规的分析思路,先对其他样本做了样本关系分析,判断样本重复性好坏,又做了差异分析,挑选出了雌雄间有显著差异的基因。
接着,对基因做了WGCNA的分析,通过模块与样本的关联分析,确定了要研究的模块,从这些与性别分化相关的模块中挑选出了连通性高的核心基因,并利用qPCR验证了他们表达量的变化,找出了他们对应调控的其他基因,构建了调控网络图。
最后结合文献,发现有一种基因在雄性分化过程中起到了重要的作用,依据这个目标基因构建了一条可能的调控通路。整篇文章依据从整体到局部再到点的思路,从样本→模块→核心基因→目标基因,最后推测了可能存在的性别决定和分化的调控机制。文章具体细节如下:

发表期刊:BMC Genomics
影响因子:3.501
合作单位:黄海水产研究所
研究背景:
虾夷扇贝(Patinopecten yessoensis)是东北亚沿海国家重要的双壳类水产经济物种,但对该物种复杂的性别分化模式的认识仍十分有限。本文筛选了与扇贝性别分化相关的基因,推测相关的调控通路。
研究材料:取雌(f)、雄(m)扇贝未成熟阶段(PyUm)、成熟阶段(PyM)、产卵期(PyO,PyS)扇贝性腺,设置6个分组:PyUmf、PyUmm、PyMf、PyMm、PyOf、PySm,每2/3/4个样品混合成一个编号样品,每组3个编号样品,对这18个样品进行RNA-seq测序。
研究思路:
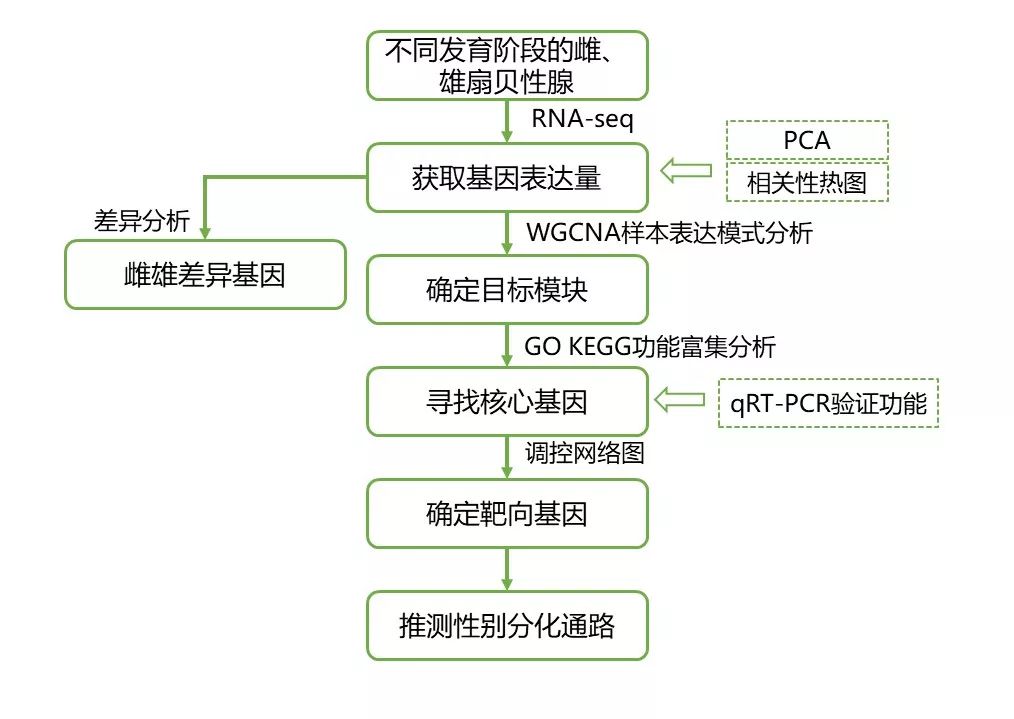
图2 研究思路
研究结果:
1. 样本相关性分析和差异分析
在实验过程中,发现PySm-1样本性腺发育异常,会影响整体的数据分析,所以剔除掉该样本进行后续主成分(PCA)分析和相关性热图分析确定其余样本重复性。确定了其余样本重复性满足要求后,对每个比较组进行差异分析,发现PyUmf vs PyUmm有3412个基因差异表达,PyOf vs PySm有2909个基因差异表达,PyMf-vs-PyMm有2778个基因差异表达(图3)。
不同阶段相同性别的扇贝相比,差异基因很少(fold change≥2,P<0.05)。对鉴定到的所有编码蛋白的基因进行分析,找到了1171个在不同性别间有显著性差异的基因,其中754个为雄性特异性基因,417个为雌性特异性基因。
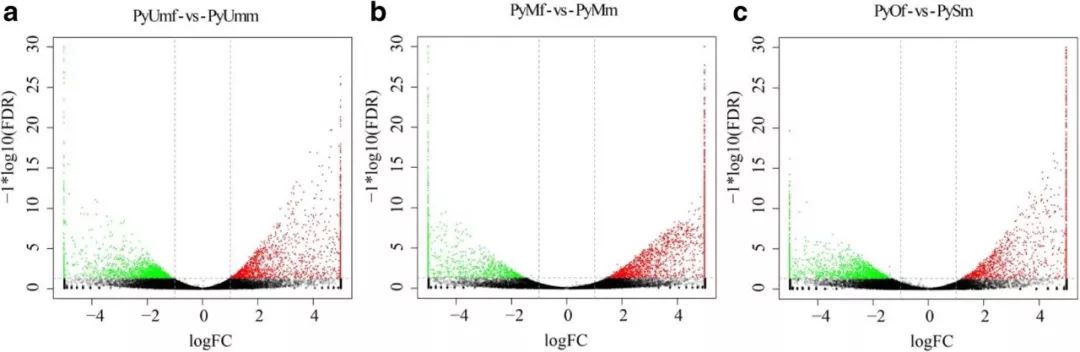
图3 三个比较组内的差异基因火山图。红色表示上调的基因,绿色表示下调的基因。
2. WGCNA分析
WGCNA分析中,模块内的基因在各个样本中的表达模式用模块特征值来展示,通过样本表达模式热图,可以找出与特定样本显著相关的模块,后续可选择相应的模块进行进一步的研究。模块特征值相当于模块中所有基因表达量的加权综合值。因此,模块特征值在各个样本中的数值,反映了模块中所有基因在各个样本中的综合表达水平。
在这篇文章WGCNA分析中,雌性和雄性扇贝的基因被划分到了19个模块。根据样本表达模式热图分析结果(图4),分别找到了与雌性和雄性显著相关的模块。与雄性性腺显著正相关的模块包括:有1541个基因的蓝绿色模块,和有1451个基因的绿色模块。与雌性性腺显著相关的模块,包括有1371个基因的coral1模块,和有860个基因的黑色模块。
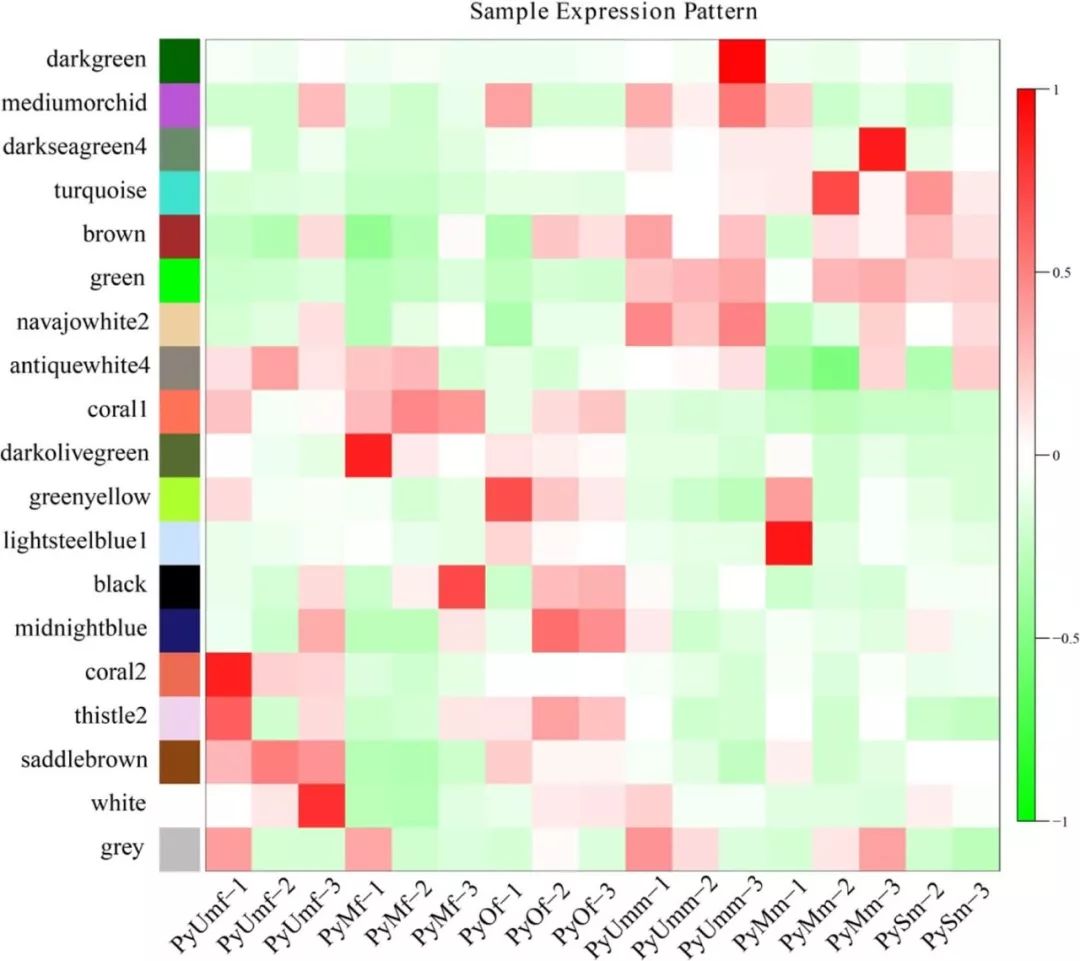
图4 样本表达模式热图分析。横坐标为样本,纵坐标为模块,用模块特征值作图。红色代表高表达量,绿色代表低表达量。该图能直观反映各模块在各个样本中的表达模式。
3. 基因功能富集分析
为了更好的理解模块内与性别相关的基因的功能,作者做了GO和KEGG富集分析(图5)。GO富集中发现大多数基因富集在biological processes这个ontology中,在五个模块中至少49个基因与性别分化或决定相关。
在KEGG的富集中,共5030个基因得到了KEGG注释。它们主要富集到代谢过程、环境信息处理功能和遗传信息加工通路。总的来看,这些基因富集到的GO term和KEGG passway与性别决定或分化相关,其中一些属于细胞过程通路,与配子发育有关。
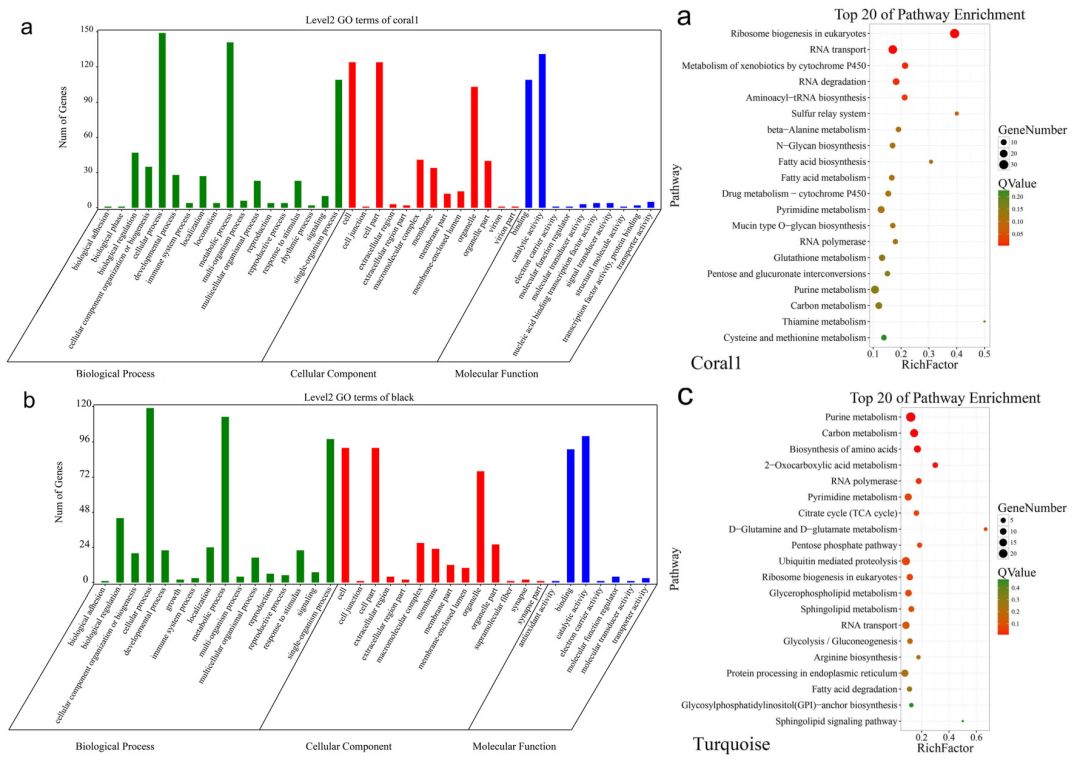
图5 GO和KEGG富集分析结果(部分)
4. QPCR验证
作者随机选择了15个性别偏倚的基因进行了qRT-PCR验证,通过将转录组数据与qRT-PCR结果比较,发现使用qRT-PCR检测的基因转录本丰度的模式与RNA-Seq结果一致。Dmrt 1(ncbi_110450487),这种基因的表达模式(图6d)和其他雄性偏倚的基因不同(图6a,b,c,e,f),与雌性偏倚的基因明显不同(图6g,h,i)。因此认为Pydmrt 1是在暗绿色(darkgreen)模块中,唯一一个在性腺早期分化过程中决定雄性性别表型的基因。
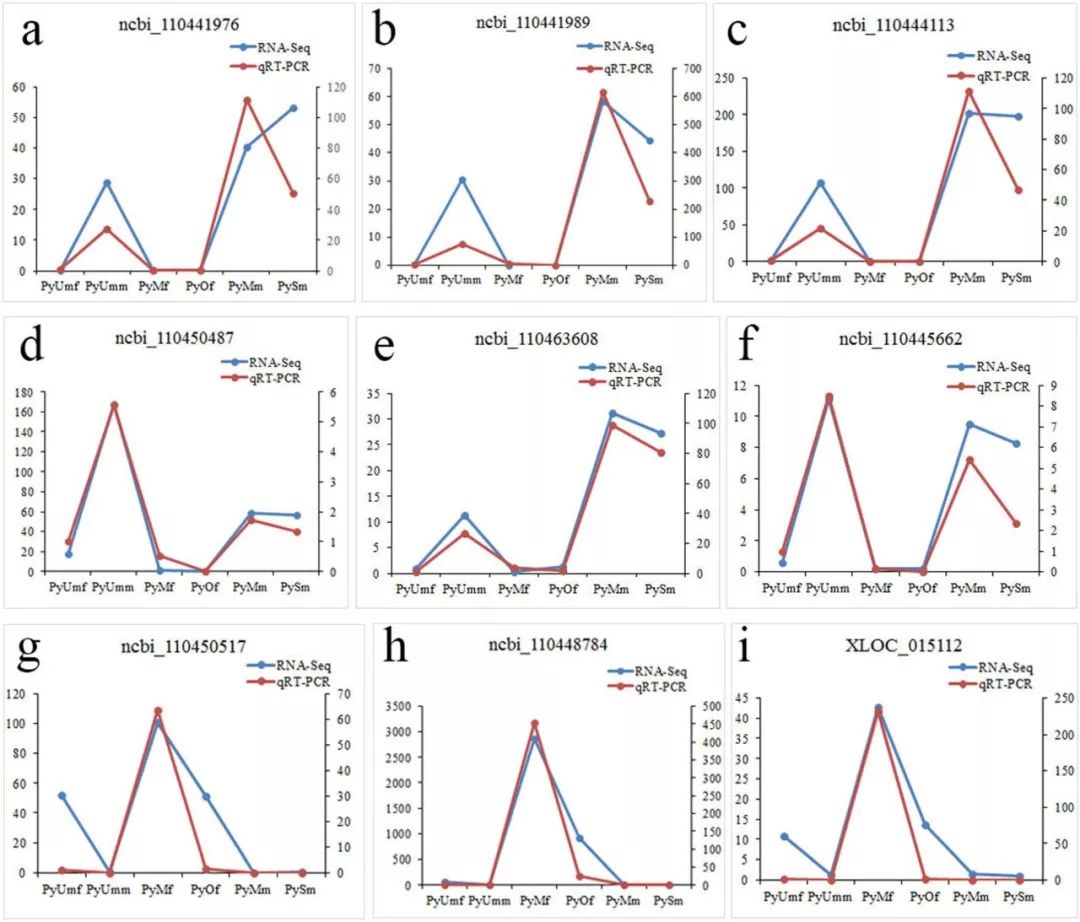
图6 对9个性别偏倚的基因进行QPCR分析和转录组表达计算。以β-actin为参照基因,用2-CT法计算9个基因的相对表达量。左边的垂直轴标度对应rna-seq值,右边的垂直轴标度对应qRT-PCR值。
5. 鉴定性别偏倚的核心基因并构建网络图
文章通过功能富集分析,在与性别分化或性别决定最相关的5个模块中,筛选出49个核心基因,根据他们在雌性和雄性组中的表达模式中的相关系数分别构建共表达网络。在雌性组中,ncbi_110450517(CYP1A4-like), ncbi_110465748(GALKNT4)和ncbi_110454866是核心基因,其他基因与这些基因正相关。在雄性组中,dmrt1是起到负调控作用的核心基因,其他是正向调控的基因(图7)。
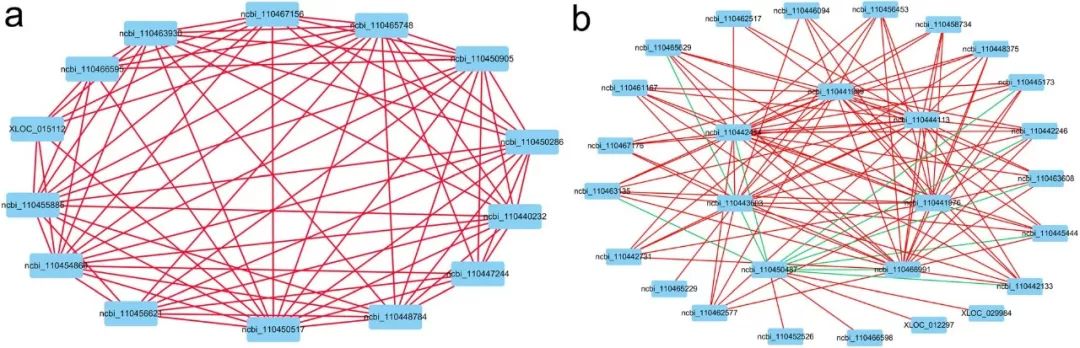
图7 利用49个基因分别构建两种性别的网络图。红线表示两种基因正相关,绿线表示两种基因负相关。a网络图依据雌性性腺转录组测序数据绘制。b 网络图依据雄性性腺转录组测序数据绘制,负调控的核心基因是ncbi_110450487(dmrt 1)。
6. 推测性别分化通路
经过qPCR验证,dmrt 1基因的表达在雄性PyUmm组中更高,在雌性所有性腺发育阶段都很低或是没有,说明Pydmrt 1的高表达在早期性腺分化阶段决定了雄性性别表型。作者推测dmrt 1基因在性别分化通路上起着主要的作用,鉴定了该基因靶向的基因,通过数据库,相关系数,表达水平,性腺发育阶段进行比较,并对他们所在的WGCNA中与性别相关的模块进行划分,最后得到了如下的性别分化通路(图8)。
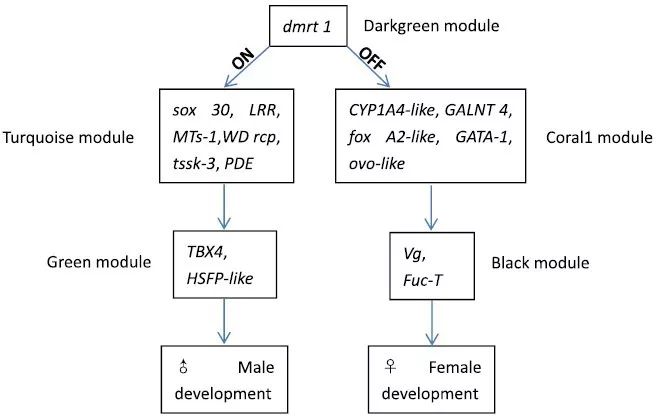
图8 推测的性别决定和分化通路
小结:
文章通过WGCNA找到了和决定性别分化的关键模块,对其中5个模块中的49个性别相关基因进行了鉴定,结合文献,确定了可能具有主导作用的Pydmrt 1基因,构建了一个可能存在的扇贝性别决定和分化通路。
在生信分析方面,文章使用了转录组常规的分析思路,在实验上只做了qPCR的验证,是研究思路比较容易复制的一类三分的文章。
如果大家想了解更多的WGCNA的知识,可以在Omicshare论坛中搜索更多的帖子进行学习,常规的转录组分析思路也可以在我们的转录组课程中进行学习,包括文章中qPCR和转录组测序结果如何通过折线图一体化展示,也可以通过我们往期的微信文章《如何一体化展示RNA-seq与qPCR定量结果》进行学习。
转自:基迪奥生物- 本文固定链接: https://maimengkong.com/zu/919.html
- 转载请注明: : 萌小白 2022年5月13日 于 卖萌控的博客 发表
- 百度已收录
