前期的推文中我们介绍了一种基于末端标签序列研究染色质交互作用的方法(ChIA-PET),本期中,我们再介绍一种应用更加广泛的染色质三维结构信息捕获方法——Hi-C.
一、Hi-C技术的发展
一提到Hi-C,我们不得不先说下它的老祖宗,3C,即Chromosome Conformation Capture。2002年,Job Dekker团队在《Science》发文,首次报道了利用Nuclear Ligation Assay的思想研究空间上相互靠近的DNA片段。由于人类基因组计划到2003年才宣告完成,所以当时还只能用半定量PCR的方法寻找具有特定反向链接的DNA片段,因此只能做到一对一。2006年,DNA芯片和测序技术推动了4C和5C的出现,做到了“一对多”和“多对多”的交互。最后,Liebermann-Aiden等在2009年开发出基于高通量测序方法在全基因组范围内研究染色质空间构象的新技术,也就是我们今天要讨论的Hi-C。
二、Hi-C方法简介
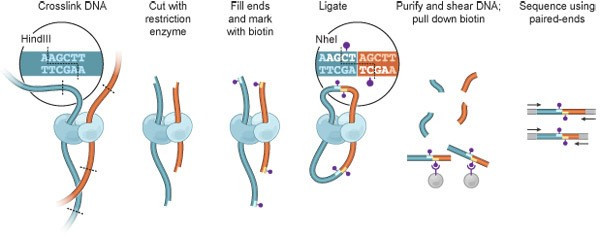
图一描绘了Hi-C技术的整个工作流程。第一步还是用甲醛使细胞内空间上靠近的DNA片段形成共价键;然后用限制性内切酶将染色质片段化;第三步用生物酰化的核酸分子链接酶切形成的粘性末端,链接过程需要在稀释的溶液中进行,有助于形成分子内链接;第四步纯化并片段化DNA,用链霉亲和素的磁珠富集含生物酰化的junction片段;最后,对收集到的junction片段进行建库并使用pair-end方法测序。
图1 Hi-C工作流程
三、Hi-C数据的生物信息学分析
Hi-C的优势在于其结合了二代测序,这势必也使得其数据分析相对复杂了。目前比较成熟的数据分析流程大致包含6个步骤:
1. 前期raw reads过滤(跟一般二代测序数据处理基本一致)
2. 序列比对。建议采用pair-end测序模式
3. 定位酶切位点。如图一所示,酶切位点的位置两端代表了相互交互的DNA片段
4. 筛选出有效的比对片段。配对的reads位于酶切位点两端且mapped的方向相反
5. 整合DNA 片段交互强度。
6. DNA片段交互矩阵标准化。
2017年6月,《Nature Methods》上发表了一篇题为《Comparison of computational methods for Hi-C data analysis》的文章,总结并比较了用于分析Hi-C数据的方法及工具,如图2.
图2,Hi-C分析方法总结

四、 Hi-C数据可视化
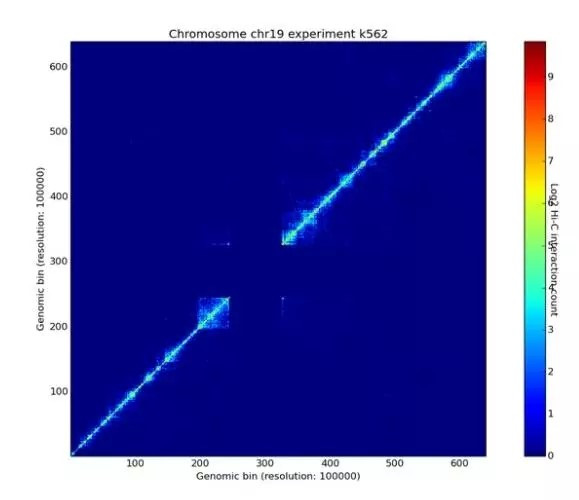
染色质DNA片段之间的交互强度以热图(heatmap)的形式表示,如图3。横轴和纵轴均表示染色质位置区间,形成二维的交互矩阵。不同的颜色代表了交互区域的强度或者显著性。Hi-C data browser网站(http://hic.umassmed.edu/welcome/welcome.php)提供帮助检索人类基因组Hi-C数据。
图3, Hi-C数据可视化热图
五、Encode 数据库中的Hi-C
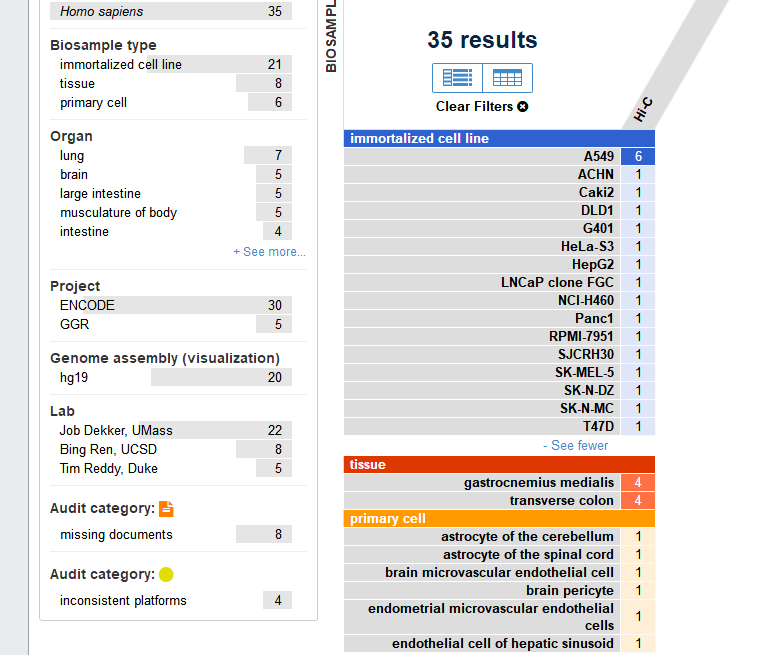
像上期一样,我们在最后给大家列出Encode数据库中已经有的Hi-C实验数据,利用这些已有的数据,找到对应于自己课题相关的细胞系或者实验设计,可以快速获得有用信息。如图4,Encode中已有35个来自人类不用组织和细胞的Hi-C数据
图4,ENCODE Hi-C数据展示
总结:基于基因组二维数据结构研究生命现象已经不能满足现在科研的需求,随着探索染色质三维结构技术的出现,能够帮助人类更深刻的理解细胞核中染色质的三维构象以及内容。我们已经介绍了ChIA-PET和Hi-C技术,但还有很多类似方法,我们会在以后的推送中陆续介绍,欢迎大家持续关注。
转自:生信草堂
- 本文固定链接: https://maimengkong.com/zu/804.html
- 转载请注明: : 萌小白 2021年11月16日 于 卖萌控的博客 发表
- 百度已收录
