整理自:https://github.com/LangilleLab/microbiome_helper/wiki/Metagenomics-standard-operating-procedure-v2
分析流程
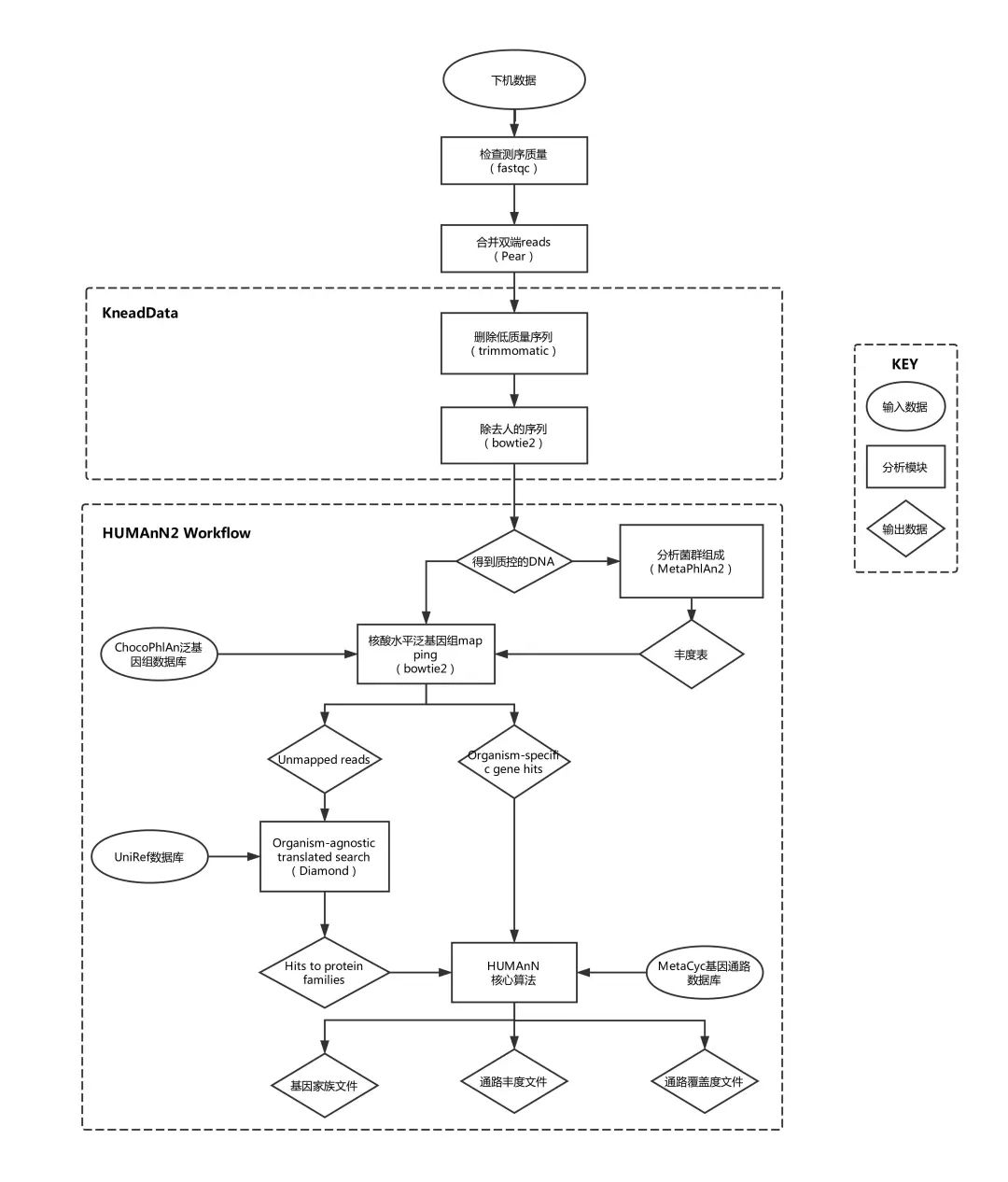
使用FastQC软件对原始数据进行质量检测,其结果包括reads各位置的碱基质量值分布、碱基的总体质量值分布、reads各个位置上碱基分布比例、GC含量分布、reads各个位置的非确定碱基数目、是否含有测序接头序列等。根据FastQC的结果,进行下一步的低质量序列的过滤及切除。
在过滤之前可使用Pear将双端测序文件进行合并操作。
使用过滤软件Trimmomatic,可以从任意一段切除低质量的碱基,同时还支持滑窗过滤,根据情况设定滑窗的大小和阈值,当滑窗内的碱基质量与设定的阈值进行比较,如果数值低于阈值则切除整个滑窗的碱基。高通量测序一般会包括接头序列以及引物片段,可以使用Trimmomatic来去除这些序列。
宏基因组测序包括了样本中所有的DNA信息,当样本取自人类粪便样本时一般会包括人类基因组的序列以及微生物的序列,所以需要去除这些人类基因组的污染序列,否则会对后续的分析造成影响,采用Bowtie2将质控后的数据与人类参考基因组进行比对,可以区分出人类和微生物的序列,从而进一步剔除这些污染序列。
获得质控的数据之后,便可进行相关样本的菌群分析,使用Metaphlan2直接将未经过拼接的测序片段比对到微生物参考数据库,从而快速获取微生物群落(细菌、古菌、真核生物和病毒)的组成,为后续的样本主坐标分析提供足够的信息。下一步,将质控的序列通过Bowtie2在核酸水平上与ChocoPhlAn泛基因组数据库进行比对,以得到泛微生物基因组的功能注释,将得到的片段再与MetaCyc通路基因数据库进行比对以得到通路信息;将上一步中未比对上的reads用Diamond加速翻译,在蛋白水平上与UniRef50数据库进行比对,以获得更多的数据。进而,通过HUMAnN2的核心算法,统计出基因的丰度信息,代谢通路的丰度信息,代谢通路的覆盖度。
所需软件
这里仅介绍所需的软件,关于各个软件如何安装,不在本文的讨论范围之内。
这里仅介绍所需的软件,关于各个软件如何安装,不在本文的讨论范围之内。
你可以在这个GitHub仓库(https://github.com/LangilleLab/microbiome_helper)得到一些用于衔接各个软件的脚本。
FastQC (可选)
官网:http://www.bioinformatics.babraham.ac.uk/projects/fastqc/FastQC是一款基于Java的软件,它可以快速地对测序数据进行质量评估(Quality Control)。
Bowtie2
官网:http://bowtie-bio.sourceforge.net/bowtie2/index.shtmlBowtie 2用于将测序reads与长参考序列进行比对,擅长于比对约50至100或1,000个字符的reads,同时也擅长与相对较大(例如哺乳动物)的基因组比对。
DIAMOND
官网:http://ab.inf.uni-tuebingen.de/software/diamond/DIAMOND用于将reads或蛋白质序列与蛋白质参考数据库(如NR)进行比对,速度高达BLAST的20,000倍,具有高灵敏性。
Human pre-indexed database
网址:ftp://ftp.ccb.jhu.edu/pub/data/bowtie2_indexes/hg19.zip人类参考基因组bowtie2的index文件,也可用bowtie2-build命令生成。
HUMAnN2
官网:http://huttenhower.sph.harvard.edu/humann2HUMAnN2是用于从宏基因组或宏转录组测序数据中高效准确地分析群落中微生物的组成在和丰度的流程,其输出文件包括基因家族文件,通路丰度文件以及通路覆盖度文件。官网:
GNU Parallel
官网:https://www.gnu.org/software/parallel/GNU parallel是处理重复命令时十分有用的工具, 这在生物信息学中尤其常用,因为我们通常希望在大批文件上运行完全相同的命令,同时以并行方式执行任务。相当于更为强大的管道xargs,推荐大家使用!
教程可参见: https://github.com/LangilleLab/microbiome_helper/wiki/Quick-Introduction-to-GNU-Parallel
教程可参见: https://github.com/LangilleLab/microbiome_helper/wiki/Quick-Introduction-to-GNU-Parallel
kneadData
官网:https://bitbucket.org/biobakery/kneaddata/wiki/HomeKneadData用于对宏基因组和宏转录组测序数据进行质量控制。在这些实验中,样品通常取自宿主,希望研究宿主上的微生物群落。 然而,来自此类实验的测序数据通常包含高比例的宿主与细菌reads。该工具旨在对来自这些“污染物”的reads分离。
MetaPhlAn2
官网:https://bitbucket.org/biobakery/metaphlan2MetaPhlAn2用于从宏基因组测序数据中分析微生物群落(细菌,古细菌,真核生物和病毒)的组成。
PEAR
官网:http://sco.h-its.org/exelixis/web/software/pear/doc.htmlPEAR会快速,高效且精确地将双端reads进行合并。
Trimmomatic
官网:http://www.usadellab.org/cms/?page=trimmomaticTrimmomatic支持多线程,处理数据速度快,主要用来去除 Illumina 平台的 Fastq 序列中的接头,并根据碱基质量值对 Fastq 进行修剪。
STAMP
官网:http://kiwi.cs.dal.ca/Software/STAMPSTAMP用于比较不同处理组之间差异物种或差异基因。该软件不仅能够对两组甚至多组样本及两两样本之间的KEGG、COG、基因及任何分类水平的物种等进行显著性差异分析,同时带有10多种可选择的差异检验方法以及图形展示形式(柱状图,散点图,热图,PCA图等)。
需要安装的Perl模块
实战
注意:
以下选项不一定适合你的数据; 探索不同的选项很重要,特别是在reads质量过滤阶段。
你应该检查每个步骤是否仅指定了正确的fastq文件(例如,使用ls命令), 如果要多次重新运行命令或使用可选步骤,则输入文件名和/或文件夹可能会有所不同。
在此工作流程中,parallel工具会多次出现。在此处查看有关此工具的教程。
建议在使用parallel时选用--dry-run选项运行,以查看正在运行的命令,以便仔细检查它们是否正在执行你想要的操作。
这份流程可用于双端测序的Miseq 或 NextSeqfastq文件,这些文件存放于raw_data文件夹。
可使用示例文件:
Step 1 (可选)将多个测序泳道数据连接在一起(例如,用于 NextSeq 数据)。如果你执行此步骤,请记住将接下来的raw_data字符更改为concat_data。
Step 2 使用fastqc检查测序质量。
fastqc参数
`-o` --outdir:输出路径
`-t` --threads:线程数
Step 3 (可选)合并双端reads(结果汇总见pear_summary_log.txt)。注意:检查组装的百分比很重要。如果reads组装的%太低,将正向和反向reads连接在一起可能会更好(见第六步)。
如果在此步骤中你没有将reads拼接在一起,则需要在运行以下命令之前解压缩fastq文件(gunzip raw_data/*gz)。
Step 4 使用kneaddata运行预处理工具。首先运行trimmomatic以删除低质量序列。然后运行bowtie2以筛选出污染序列。接下来,筛选出映射到人类基因组的reads。注意,在这里我们用parralel对kneaddata进行并行运算,点击这里看到关于这个工具的快速教程。有关以下命令中的选项的详细介绍,请参阅此页面。正向和反向reads将由输出文件中的_1和_2指定。注意,每行末尾的\只是将命令分成多行,以便于阅读,没别的意思。
parallel参数
-j/--jobs:并行任务数
kneaddata参数
-i 为输入fastq文件,双端需输两次
-o 输出结果目录
-db 指定bowtie2索引
--trimmomatic 指定质控程序位置
--trimmomatic-options 质控选项,"SLIDINGWINDOW:4:20 MINLEN:50"表示:4碱基滑窗,质量大于20,最小长度50
-t 设置线程数
--bowtie2-options 比对选项
--remove-intermediate-output 清理中间文件
你也可以用以下命令生成日志文件:
Step 5 (可选)如果你担心影响样本中测序深度的差异,参阅此页面可能会有所帮助。
Step 6 (可选)如果你没有用pear进行合并双端reads,那么你现在可以将正向和反向的fastq连接在一起。请注意,这是在质量过滤后完成的,因此双端测序中的两个reads都将被一起丢弃或保留。注意,我们只需要指定kneaddata输出的“配对”的fastq文件。你需要先将污染物的序列移动到单独的文件夹中(用下面前两个命令)。
Step 7 用parallel并行运算humann2来计算uniref90基因家族和metacyc通路的丰度。
报错:The database file for MetaPhlAn does not exist at /data/zwbao/biosoft/metaphlan2/db_v20/mpa_v20_m200.pkl . Please provide the location with --metaphlan-options .
解决方法:cd到metaphlan2安装目录下的db_v20文件夹中(若没有这个文件夹则新建一个),在 https://bitbucket.org/biobakery/metaphlan2/downloads/ 下载mpa_v20_m200.md5和mpa_v20_m200.tar文件,解压mpa_v20_m200.tar得到.fna和.pkl。
报错:The database file for MetaPhlAn does not exist at /data/zwbao/biosoft/metaphlan2/db_v20/mpa_v20_m200.pkl . Please provide the location with --metaphlan-options .
解决方法:cd到metaphlan2安装目录下的db_v20文件夹中(若没有这个文件夹则新建一个),在 https://bitbucket.org/biobakery/metaphlan2/downloads/ 下载mpa_v20_m200.md5和mpa_v20_m200.tar文件,解压mpa_v20_m200.tar得到.fna和.pkl。
Step 8 将每个样本的humann2输出导入到一个表中。
Step 9 重新标准化基因家族和通路的丰度(标准化为百分比)。
Step 10 分割humann2输出的丰度表为分层(unclassified)和非分层表。
Step 11 通过改变标题行将未分层的humann2丰度表转换为STAMP格式。这些命令会删除注释字符和第一列名称中的空格。每个样本的列名也会被删除一些描述。
Step 12 因为humann2也运行metaphlan2作为初始步骤,我们可以使用已经创建的输出表来获得我们样本的分类组成。首先,我们需要将每个样本的metaphlan2结果收集到一个目录中,然后使用metaphlan2的merge_metaphlan_tables.py命令将它们合并到一个表中。之后,我们删除_metaphlan_bugs_list这串字符。
Step 13 最后我们将这个metaphlan2丰度表转换为STAMP格式。结果可使用STAMP打开,进行常见统计分析的可视化,点此查看软件主页。
metaphlan_to_stamp.plmetaphlan2_merged.txt > metaphlan2_merged.spf
转自生信草堂公众号
- 本文固定链接: https://maimengkong.com/zu/764.html
- 转载请注明: : 萌小白 2021年8月25日 于 卖萌控的博客 发表
- 百度已收录
