MaxQuant是一款免费的蛋白质组学软件,它内置了自己的搜索引擎Andromeda,可以支持目前主流的蛋白质组学质谱仪厂商产生的原始数据格式(raw,wiff,mzML,mzxml)。
MaxQuant支持标记定量和非标定量,2.0版本后的MaxQuant还支持DIA的数据分析。MaxQuant具备非线性质量校正和Match Between Runs功能,可以增加蛋白鉴定数量和提高定量准确性。
01 下载软件
进入官网maxquant.org ,选择MaxQuant软件,解压后无需安装,点击MaxQuant.exe即可使用
02 下载待搜库物种蛋白质序列
示例如下,下载人类蛋白组序列

打开UniProt网站,选择Taxonomy 》human
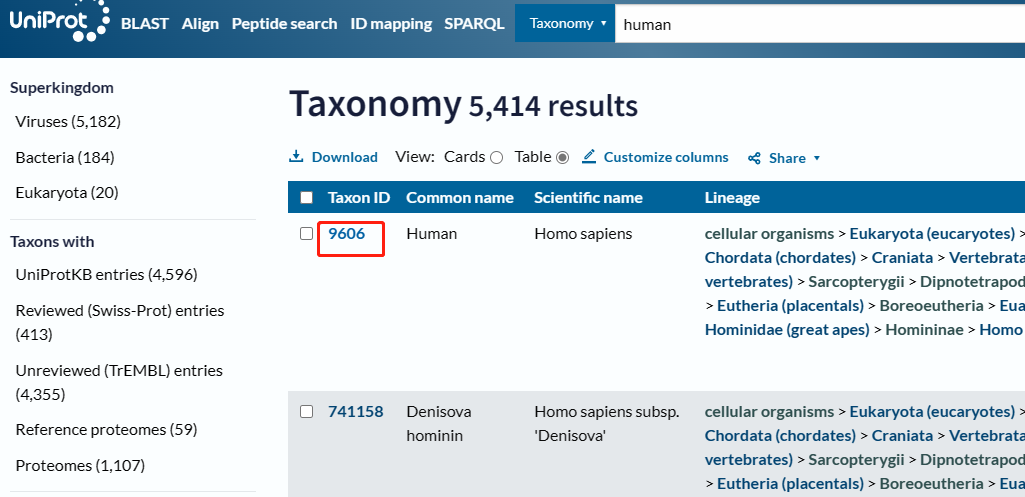
点击taxid 9606

选择View proteomes中的 Reference proteomes
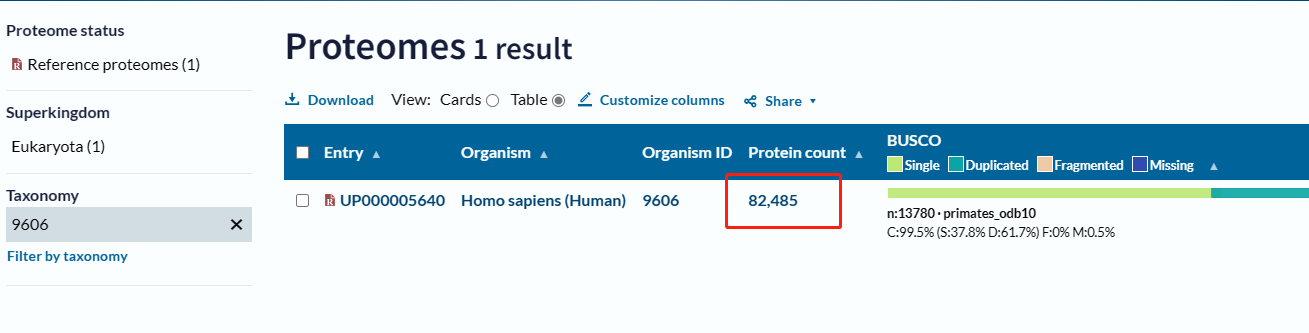
点击该数据库含有的蛋白数目82485
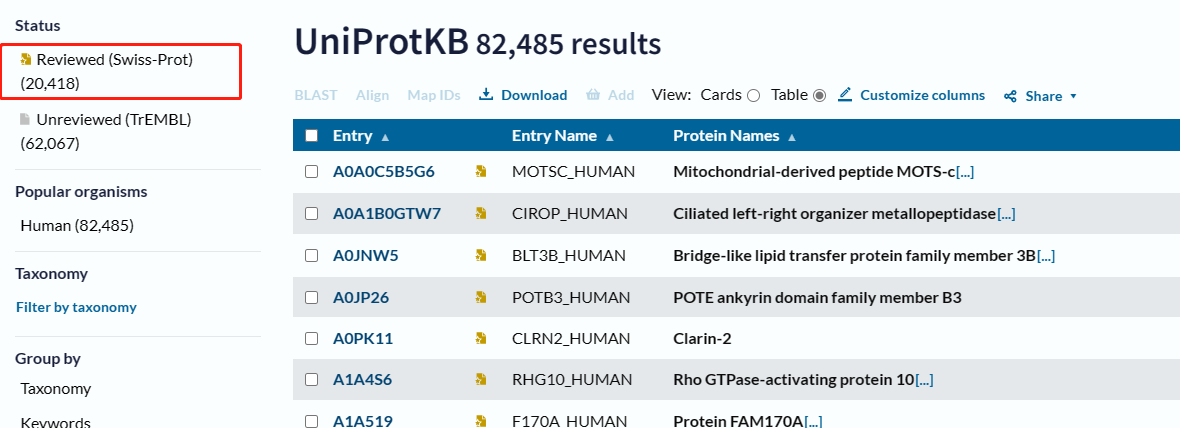
点击左上角的Reviewed (Swiss-Prot),它是Reviewed,即人工审阅,归属于UniProt下的Swiss-Prot数据库,选中后点击右侧的Download即可
03 使用MaxQuant(示例软件版本:MaxQuant_2.4.7.0)
MaxQuant对蛋白质序列数据库fasta文件中的蛋白质序列进行模拟酶切和碎裂,将蛋白质序列信息转换为理论谱图数据。通过将理论谱图数据与实际采集到的原始谱图数据进行比对打分,实现搜库的过程。
03-1 导入数据
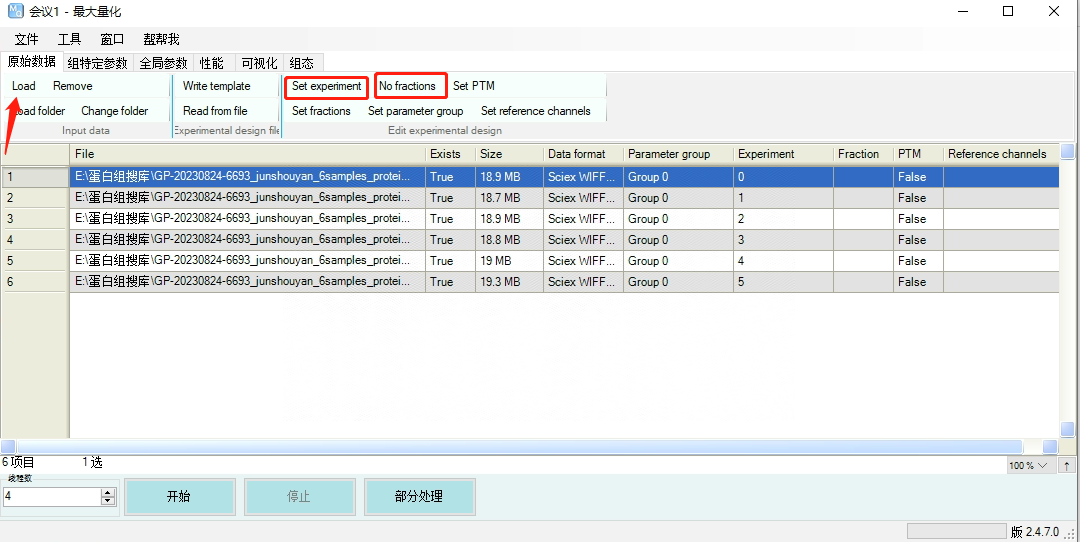
Load导入质谱数据,主要原始数据格式有.raw (Thermo公司)、.d (Bruker公司)、.WIFF和.wiff.scan (SCIEX公司)等。
Set experiment设置实验组名称,注意如果设置相同的名称,后续搜库时会被合并。
或样本数量多时可以像截图中一样直接点击 No fractions 自动导入0-5(Experiment列)的数字,后续可在搜库结果里修改。
03-2 设定组特定参数
对于常规的非标定量检测,可直接使用软件默认的参数
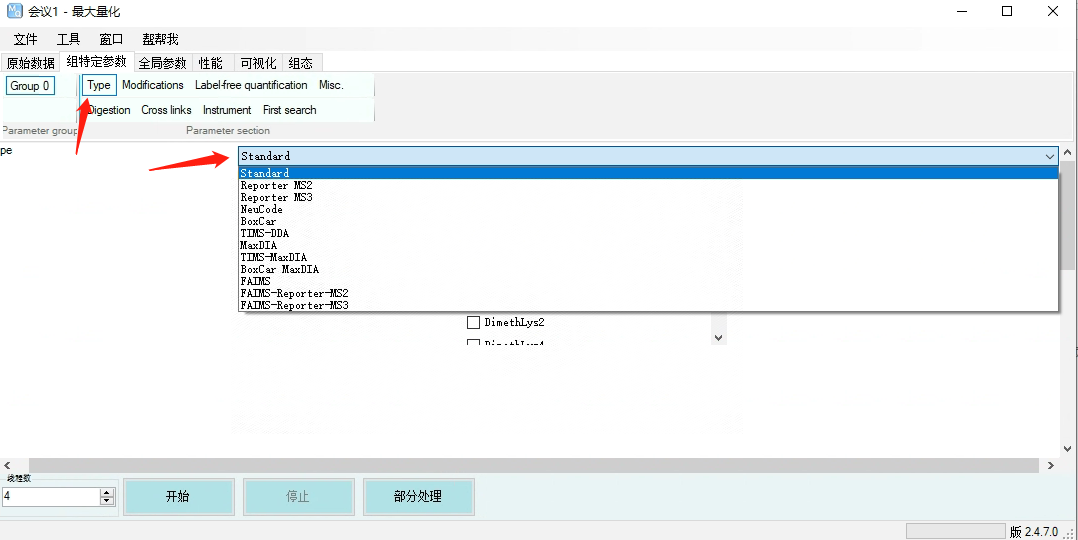
Type——默认选择Standard,label-free和MS1-labeled样本选择Standard,传统的同位素标记标记样本(TMT)选择Reporter ion MS2,同位素标记的MS3质谱选择Reporter ion MS3
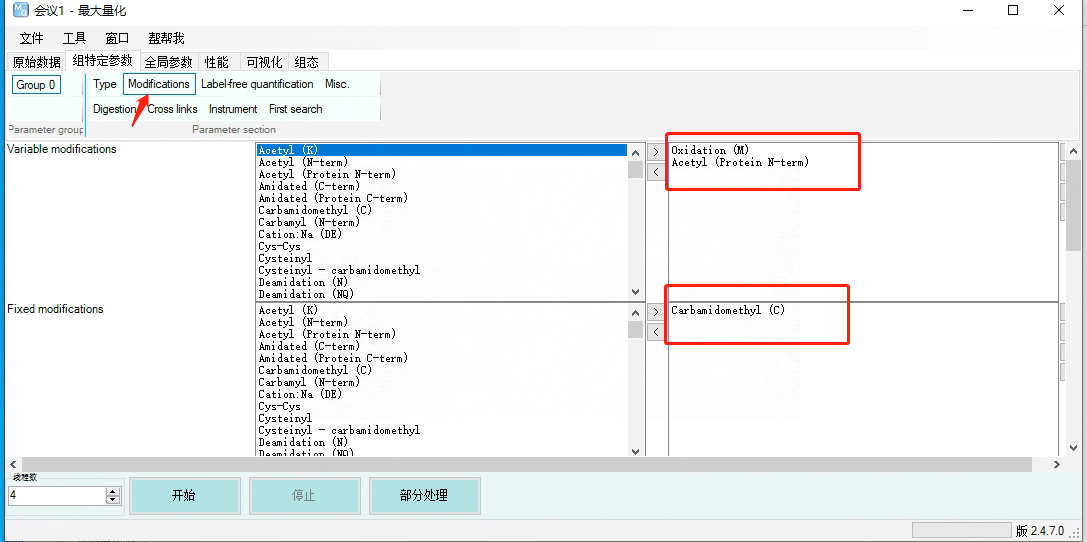
Modifications——默认选择固定修饰Carbamidomethyl (C)、可变修饰Oxidation (M)和Acetyl (Protein N-term)
Variable modifications是指并非每个氨基酸都会发生的修饰,如蛋氨酸氧化(Oxidation (M))可能只发生在某些蛋氨酸上。再比如,只有少数肽的 N 端会发生乙酰化(Acetyl (Prorein N-term))。需要注意的是可变修饰会增加in silico肽数据库的容量,进而增加分析时间。
Fixed modifications是指特定氨基酸每次出现时都会发生的修饰。这些修饰通常是人为引入的,如蛋白质组学样品制备中防止二硫键重新形成的半胱氨酸的氨基甲酰甲基化,也是该参数的默认值Carbamidomethyl (C)。
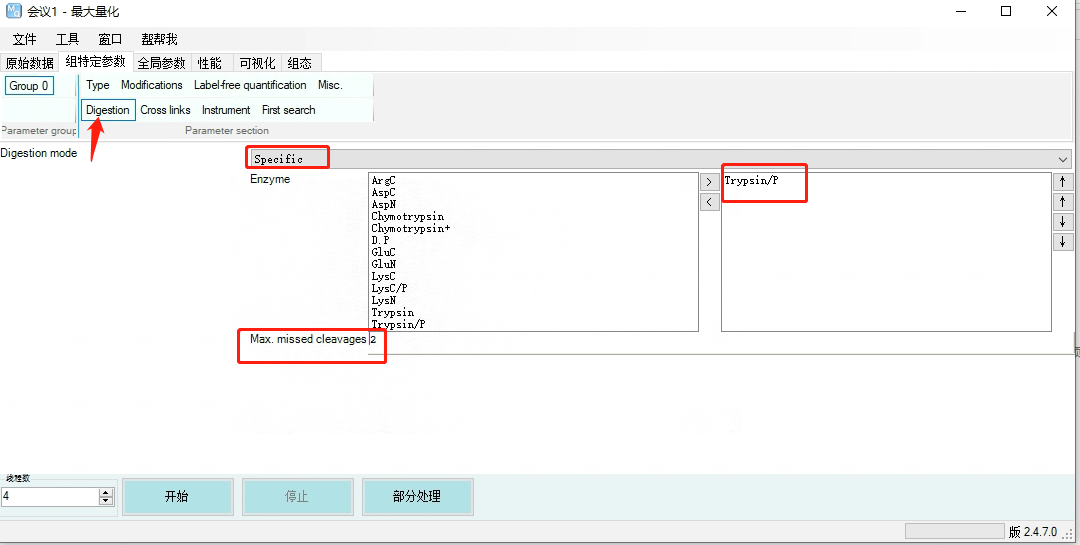
Digestion——默认选择Specific和Trypsin/P。Max. missed cleavages默认为2。
因为准确的裂解特异性等优点,胰蛋白酶在多数自下而上的蛋白质组学实验中都被用作蛋白酶。因此Enzyme默认值为Trypsin/P。胰蛋白酶能裂解精氨酸和赖氨酸C末端的肽段,除非其后有脯氨酸。MaxQuant使用该规则对蛋白质数据库进行in silico裂解。
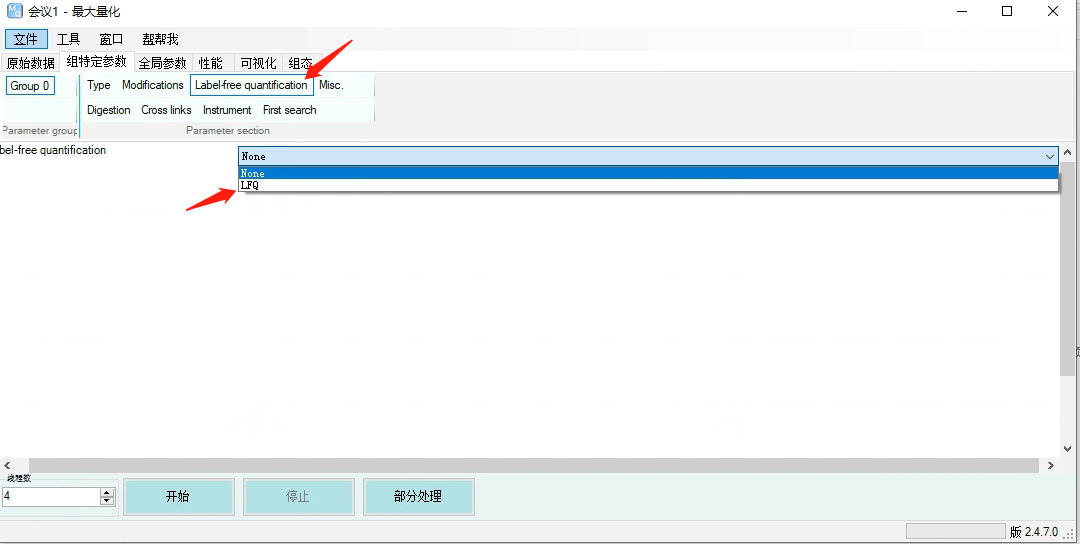
需要修改的参数:Label-freequantification——选择LFQ。
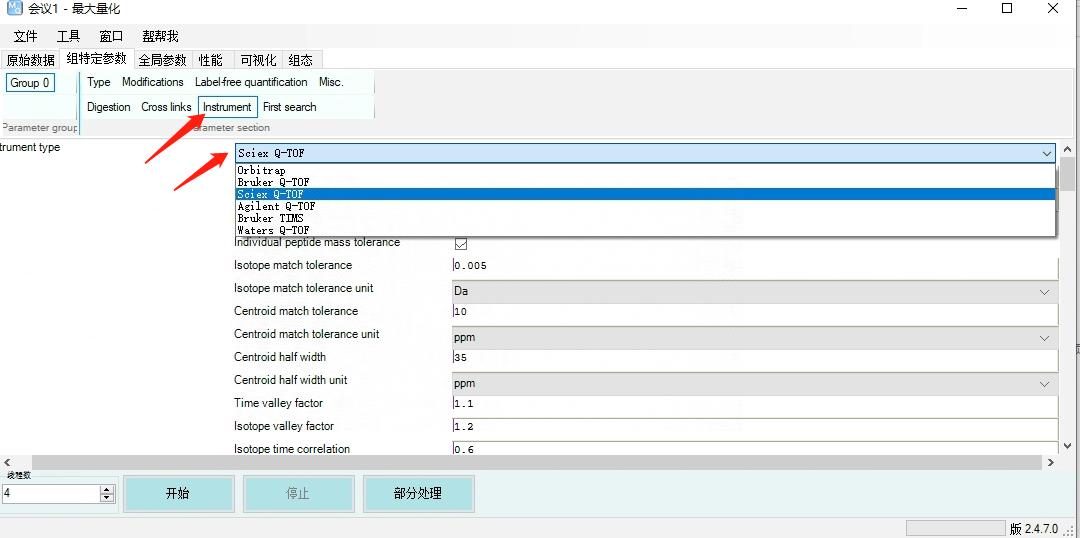
需要修改的参数:Instrument——根据实际质谱检测的仪器选择Instrument type
03-3 设定全局参数
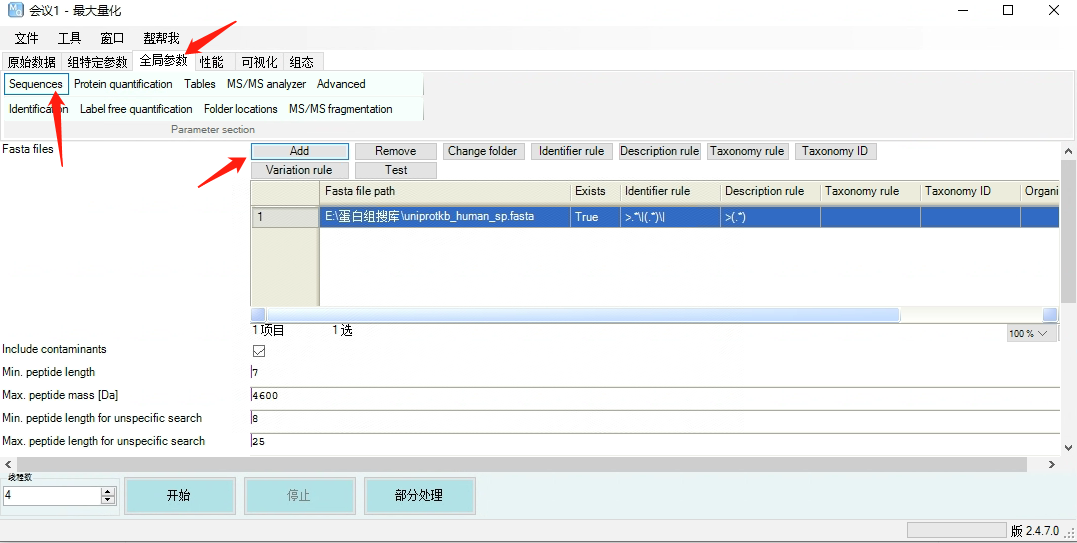
Sequence——add导入参考的Human蛋白质序列数据库fasta文件
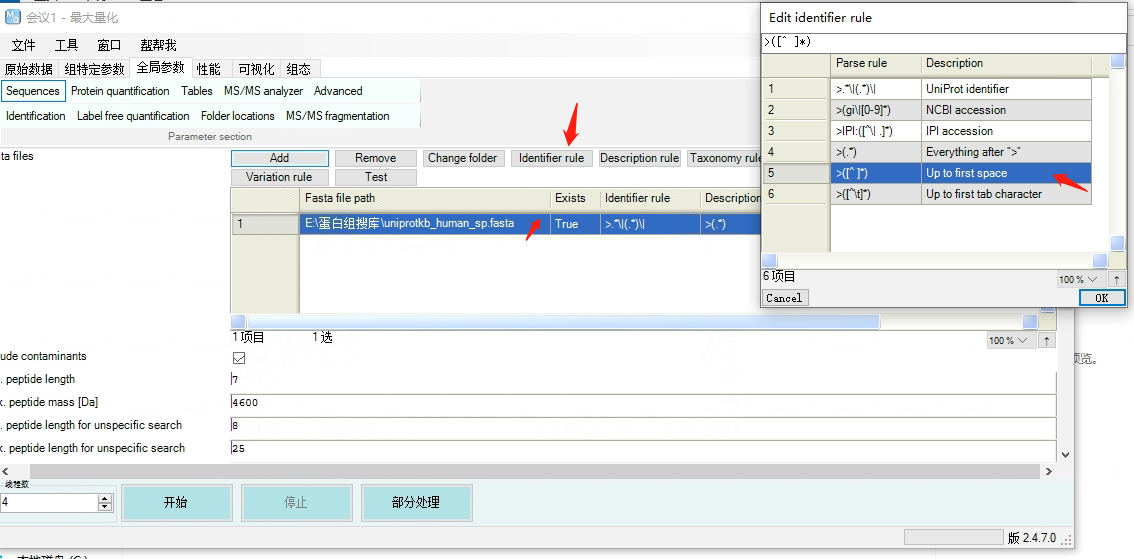
选中蛋白序列行,点击Identifier file ,选中第5个Up to fisrst space ,点击ok
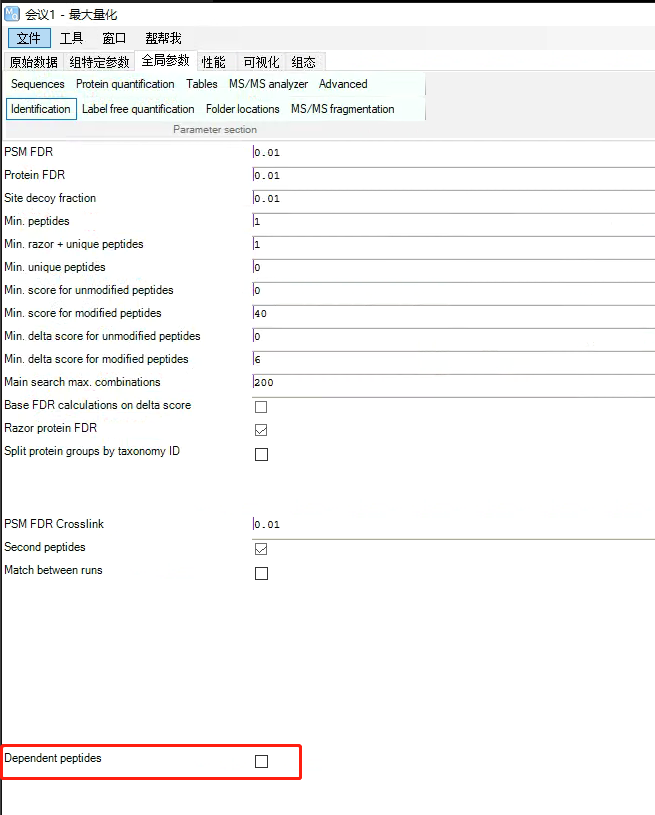
可勾选Identification——Match between runs。
由于缺失值的存在会削弱生物样本或实验条件之间真实定量差异的能力,Match between runs功能可以改善缺失值问题。
PSM FDR:PSM水平的FDR阈值,由TDA计算得出,0.01代表1% FDR。
Protein FDR:蛋白质水平的FDR阈值。
Site decoy fraction:修饰位点表水平上对饵命中进行过滤。
Min. peptides:鉴定一个蛋白质簇所需总肽段数的最小值,并在最终表格中报告。
Min. razor + unique peptides:鉴定一个蛋白质簇所需razor+unique肽段数的最小值,并在最终表格中报告。
Min. unique peptides:鉴定一个蛋白质簇所需unique肽段数的最小值,并在最终表格中报告。肽会以统计的方式组合成相应的蛋白质,因此主要应根据唯一肽进行判断。可以将参数设为1,即输出表中只报告至少有一个唯一肽的蛋白质。
Min. score for unmodified peptides:接受MS2鉴定无修饰肽的最小Andromeda分数。推荐值为0,即除FDR外,没有额外的过滤。
Min. score for modified peptides:接受MS2鉴定修饰肽的最小Andromeda分数。默认值为40。除对PSMs应用的FDR外,还需额外过滤。
Min. delta score for unmodified peptides:接受MS2鉴定无修饰肽的最小Andromeda delta分数。推荐值为0,即除FDR外,没有额外的过滤。
Min. score for modified peptides:接受MS2鉴定修饰肽的最小Andromeda delta分数。默认值为6。除对PSMs应用的FDR外,还需额外过滤。
Base FDR calculations on delta score:如果选中,所有 FDR 计算都将使用 delta 分数作为输入。delta 分数是得分最高的 PSM 与氨基酸序列不同、得分次之的 PSM 之间的分数差。
Dependent peptides:无偏搜索来自已鉴定肽的修饰肽。
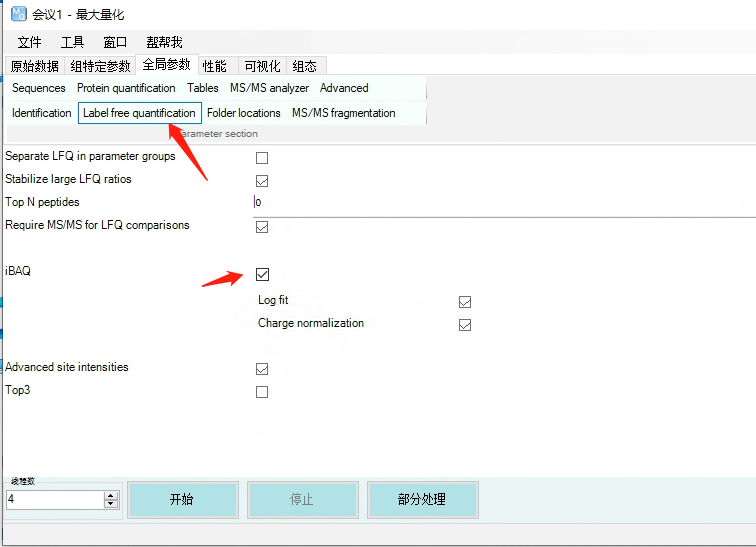
Label-free quantification——勾选iBAQ。iBAQ是基于Intensity的强度值,除以该蛋白的理论可被检测的肽段数目计算而来的定量值,主要用于不同蛋白的相互比较。
03-4 设置线程数,运行
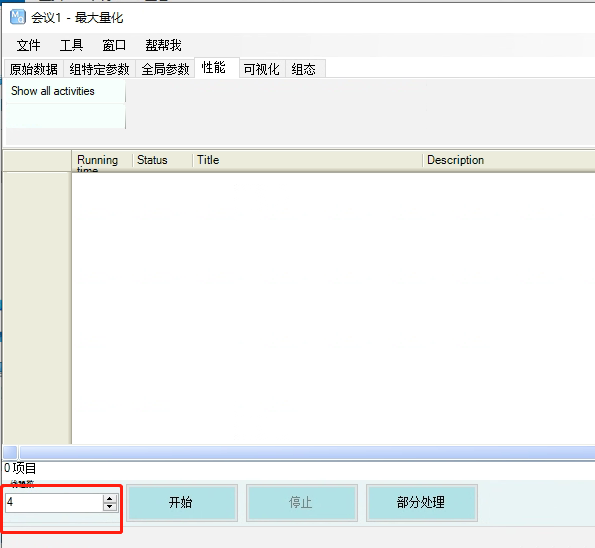
可以根据电脑的cpu数量进行设置
04 搜库结果文件展示
- 本文固定链接: https://maimengkong.com/zu/1689.html
- 转载请注明: : 萌小白 2024年2月13日 于 卖萌控的博客 发表
- 百度已收录
