在单细胞研究中,最重要的一个环节就是细胞类型注释,这是一个极其考验研究者研究背景和精力的工作,但随着单细胞的研究越来越多,可提供给我们的细胞类型的marker信息也越来越丰富,基于这些marker信息开发的细胞注释算法使得我们的细胞注释工作越来越省力,今天就跟随小编的脚步来盘点一下最常用的细胞类型注释工具吧!
SingleR:
最早是来自一篇肺部巨噬细胞研究的单细胞论文,这篇文章作者用多份实测数据证明了SingleR软件可以较好基于单细胞转录组数据对各个细胞进行鉴定。该算法基础的工作原理很简单:准备一套参考数据集,参考数据中每个样品被人工注释为一种主要的细胞类型,及相应的细胞亚型标签;然后通过差异表达的方法或方差分析的方法获取到已知细胞类型的variable genes,在variable genes 中计算每一个单细胞与参考数据集中每一个样品的spearman相关系数,同一细胞类型下多个参考样品的相关系数的80%分位数作为这个单细胞注释到此细胞类型的得分;保留与参考细胞类型注释最大得分差值在0.05以内的参考细胞类型及,重新计算 variable genes,再次计算测试细胞与剩下参考细胞类型集的相关系数,迭代,直到只剩下两种细胞类型时,保留相关性得分最高的已知细胞类型,为此细胞注释到的细胞类型。
目前SingleR内置的数据库有7个,5个人的数据库HumanPrimaryCellAtlasData、BlueprintEncodeData、DatabaseImmuneCellExpressionData、MonacoImmuneData、NovershternHematopoieticData;2个小鼠的数据库ImmGenData、MouseRNAseqData,可应用于相应物种及组织的单细胞结果注释。
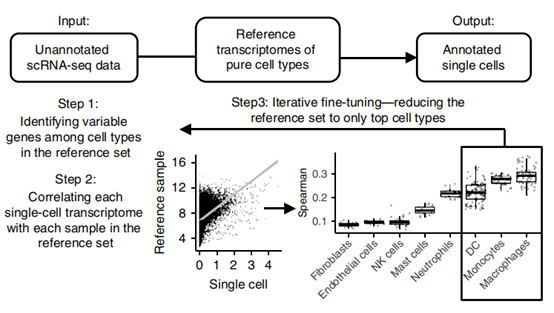 SingleR单细胞注释工作原理
SingleR单细胞注释工作原理
Cellassign:
基于marker基因的信息自动将单细胞RNA-seq数据分配注释到已知的细胞类型中。它以marker基因的细胞类型矩阵作为输入,提供先验的已知marker基因是否属于某种细胞类型,然后,Cellassign会概率性地将每个细胞分配给一个细胞类型,从而消除了典型无监督聚类中的主观偏见。该方法开发初衷是应用于肿瘤微环境的分析,但由于其对细胞类型的注释依赖于marker列表,因此应用非常广泛,并且理论上是没有明确的物种限制。
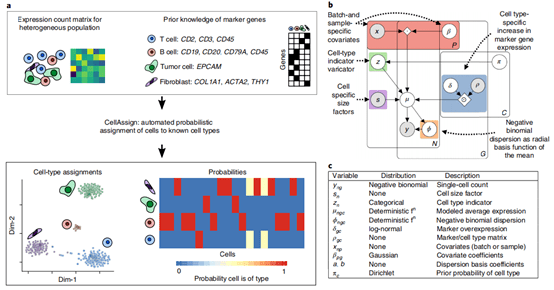 Cellassign单细胞注释工作原理
Cellassign单细胞注释工作原理
Celaref:
通过与已知细胞类型的参考数据集的相似度进行比较。输入每个细胞中每个基因的readscounts数(gene-cell matrix)和每个细胞所属的簇(cluster)信息,和每个查询组中最明显富集的基因的参考样本比较,通过排名来匹配细胞类型,该算法主要使适用于人和小鼠。
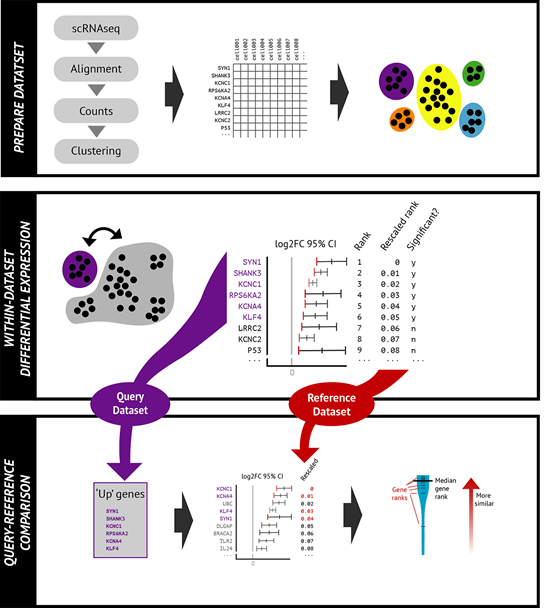 Celaref单细胞注释工作流程
Celaref单细胞注释工作流程
Garnett:
是一个从单细胞表达数据中实现自动细胞类型分类的软件包。Garnett的工作方式是获取单细胞数据和细胞类型定义(marker)文件,并训练一个基于回归的分类器。一旦被训练成一个针对某一组织/样本类型的一个分类器,它就可以应用于从相似组织中对未来的数据集进行分类。除了描述训练和分类功能,这个网站的另一个目标是成为一个存储以前训练出来的分类器仓库,该算法已经为各种生物和组织生成了一系列预先训练的分类器,包括人,小鼠,线虫的多种组织,如果我们使用的物种没有可用的AnnotationDbi类数据库,则Garnett将无法在基因ID类型之间进行转换,但是我们仍然可以使用Garnett进行细胞分类。
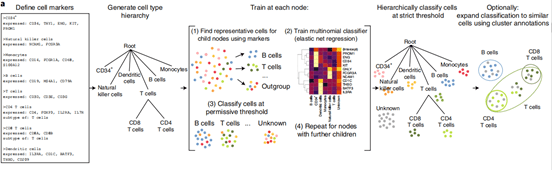 Garnett单细胞注释工作原理
Garnett单细胞注释工作原理
scTPA:
是用于在人和小鼠中基于生物通路激活而进行单细胞转录组分析和注释的网络工具。数据库收集了具有不同功能和分类的大量生物通路,这有助于识别细胞类型和解释关键通路特征。优化了四种不同的通路激活评估方法的可执行代码,以使运行时间减少4至56倍。提供了单细胞通路激活概况的分析和可视化功能,例如细胞聚类和注释,标记通路及其相关基因的鉴定,从面向通路的角度,这将有助于更好地了解细胞类型和状态。
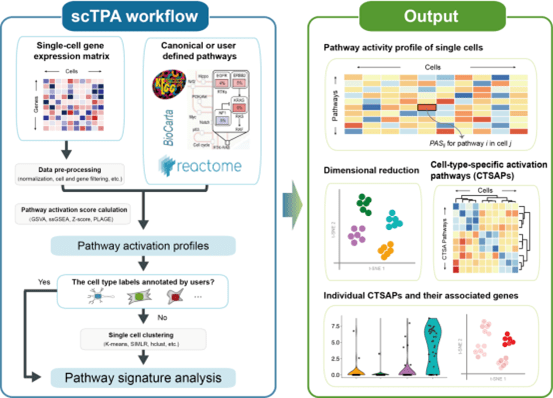 scTPA单细胞注释工作原理
scTPA单细胞注释工作原理
Cell Blast:
该算法由北京大学生物医学前沿创新中心(BIOPIC)、北京未来基因诊断高精尖创新中心(ICG)、北京大学生命科学学院生物信息中心(CBI)、蛋白质与植物基因研究国家重点实验室的研究团队基于深度学习模型开发的scRNA-seq数据检索和注释的新方法,Cell BLAST可以在参考数据集中检索与用户提供的query细胞最相似的细胞,并借助这些相似细胞在数据库中的注释信息,对query细胞的注释信息进行推断。该算法自带高质量注释的scRNA-seq参考数据库Animal Cell Atlas (ACA),是一个涵盖2,989,582个单细胞、8个物种、27个不同的组织器官的数据库,这一数据库为有效利用现有数据进行细胞注释和跨数据集研究提供了新的工具和资源。
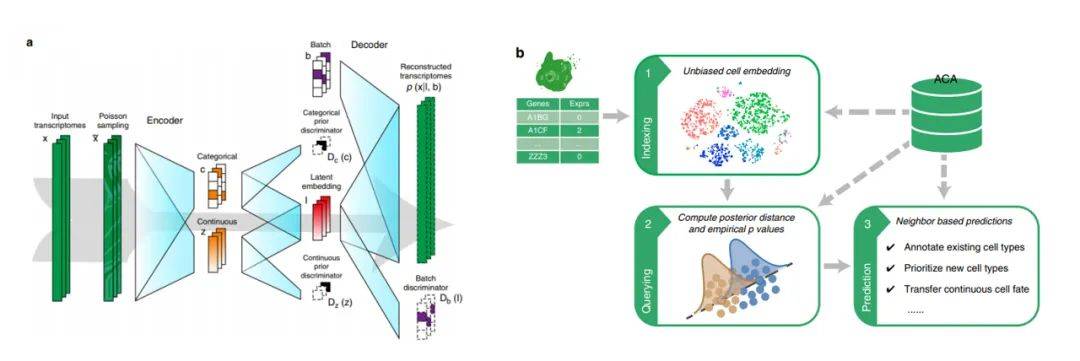 Cell Blast单细胞注释工作原理
Cell Blast单细胞注释工作原理
scCATCH:
由浙江大学药学院药物信息学研究所研究团队开发,全称是single cellCluster-based Annotation Toolkit for Cellular Heterogeneity,是一个用于实现单细胞转录组聚类结果进行注释的工具。研究结果表明使用该方法在6种不同组织(Pancreas、Brain、Lung、PBMCs、Brain和Brain)的测试数据中的平均标注准确率为83%。方法主要包括两个函数“findmarkergenes”和“scCATCH”,以实现对每个已识别集群的自动注释,该算法内置数据库CellMatch包含353种细胞类型和686种亚型,184种组织类型,20,792种细胞特异性标记基因以及2,097个人类和小鼠参考文献,数据库中人的组织和肿瘤以及小鼠的正常组织的参比资源很多,只适用于人和小鼠的单细胞数据注释。
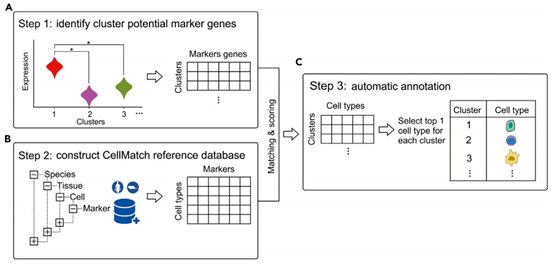 scCATCH单细胞注释工作原理
scCATCH单细胞注释工作原理
- 本文固定链接: https://maimengkong.com/zu/1520.html
- 转载请注明: : 萌小白 2023年5月6日 于 卖萌控的博客 发表
- 百度已收录
