1. CircRNA背景及分析流程简介
1.1. 背景简介
环形RNA是一类在真核生物中广泛存在的具有特殊环状结构的非编码RNA分子。已有文献表明,在生物体内,环形RNA有着miRNA海绵、RBP海绵以及翻译短肽等多项功能,在许多生物学过程中发挥着重要作用。
目前研究表明,大部分环形RNA来源于蛋白编码基因的外显子区域。在pre-mRNA剪接的过程中,除典型的内含子剪接事件外,还可能会发生5’端到3’端的反向剪接事件,从而形成环形RNA。因此,剪接产物中环形RNA所占比例是环形RNA分析的重要指标之一,具有高成环比例的环形RNA分子,可能具有更加重要的生物学功能。
同时,同一基因内部也可能产生多种不同的环形RNA,基因内对环形RNA产生位点的使用偏好,也在一定程度上反映了转录过程对环形RNA产生的调控。因此,环形RNA转录本水平的准确定量,是目前环形RNA分析的重要基础。
为了解决该问题,赵方庆团队开发了一个新的环形RNA分析算法。根据已有工具鉴定出的环形RNA成环位点信息,研究人员重构了具有反向剪接特征的环形RNA参考序列,简化了复杂的反向剪接位点比对问题,并结合测序读段比对到参考基因组和环形序列的结果,筛选出了高置信度的来自环形RNA的读段,解决了目前环形RNA识别和定量方法中准确度低和假阳性率高的问题。作者在模拟数据和真实转录组数据中,对多种常用环形RNA识别软件的表现进行了综合评估,发现该研究中开发的方法在环形RNA表达量和成环比例的计算中,均取得了最佳的结果。
1.2. 分析流程
将下机测序数据进行质控,去除接头及各类低质量序列。随后借助于CIRIquant,使用Hisat2与参考基因组比对,Stringtie进行基因水平定量;同时使用bwa-men与参考基因组比对,进行circRNA的鉴定,构建circRNA参考序列;将构建的circRNA序列作为参考基因组使用Hisat2再次进行比对,筛选出高置信度的来自环形RNA的reads;统计circRNA的表达情况,并注释circRNA信息。通过对circRNA差异分析,筛选出具有显著差异的circRNA所对相应的基因进行后续富集分析。 circRNA信息分析简易流程如下所示。
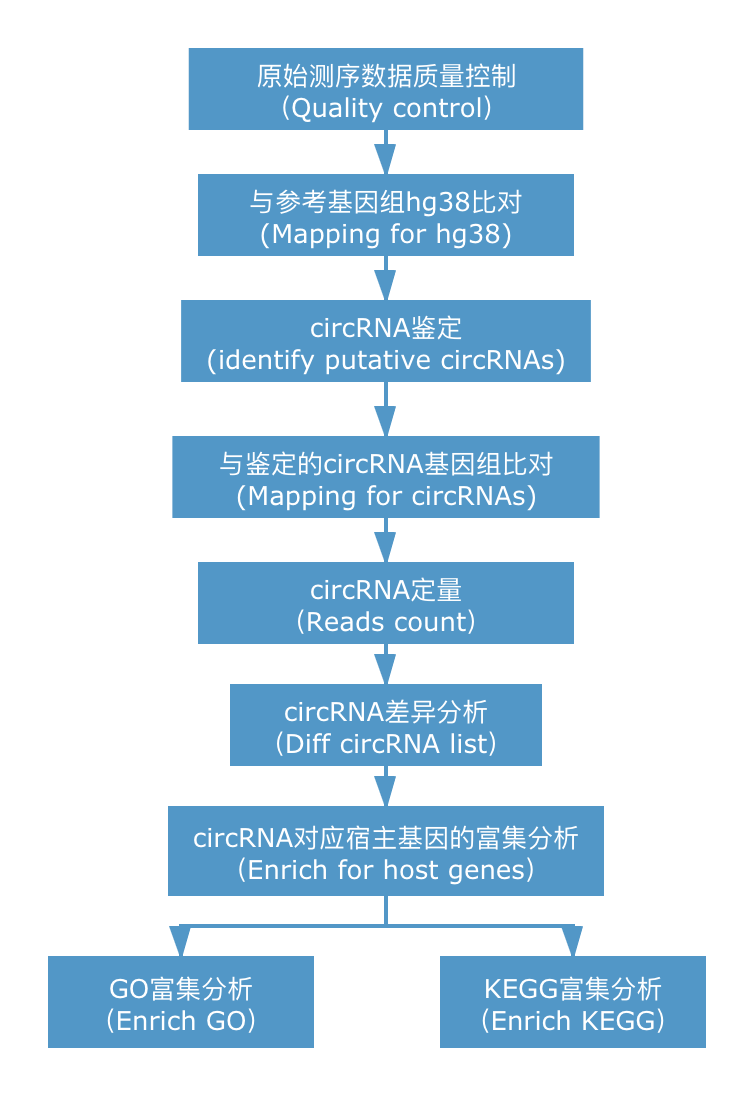
2. 分析结果
2.1. 测序数据质量控制
对原始测序数据及去除接头后的可用数据进行质量评估。测序数据一般为双端测序,因此,每个测序样本会有两个测序结果。
评估的具体内容:
| 文件路径 | 文件说明 |
|---|---|
result/01.QC/qc_rawdata/*.html
|
RawData-fastqc 文件链接 |
result/01.QC/qc_cleandata/*.html
|
CleanData-fastqc 文件链接 |
result/01.QC/qc_Supplement.html
|
Fastqc 格式补充说明 |
2.2. CIRIquant分析
2.2.1. CIRIquant分析结果文件
1 . 比对结果文件:
| 结果路径 | 结果说明 |
|---|---|
result/02.CIRIquant/1.mapping/*.flagstat.txt
|
各个样本的比对情况统计 |
result/02.CIRIquant/1.mapping/*.bw
|
与hg38基因组比对的bw结果 |
以上结果位于文件夹:result/02.CIRIquant/1.mapping/
2 . CIRIquant鉴定结果文件:
| 结果路径 | 结果说明 |
|---|---|
result/02.CIRIquant/2.circRNA_detection/*.gtf
|
CIRIquant鉴定circRNA的gtf文件 |
result/02.CIRIquant/2.circRNA_detection/*.bed
|
CIRIquant鉴定circRNA的bed文件 |
以上结果位于文件夹:result/02.CIRIquant/2.circRNA_detection/
3 . CIRIquant鉴定结果的统计结果文件:
| 结果路径 | 结果说明 |
|---|---|
result/03.circRNA_info/1.circRNA_annotation/*csv
|
鉴定的circRNA的注释信息表 |
result/03.circRNA_info/2.circRNA_length/*
|
鉴定的circRNA的长度分布图 |
result/03.circRNA_info/3.circRNA_karyotype/*
|
鉴定的circRNA的染色体分布图 |
result/03.circRNA_info/4.circRNA_type/*
|
鉴定的circRNA的类型统计图 |
以上结果位于文件夹:result/03.circRNA_info
以上统计图的可视化文件:result/03.circRNA_info/view.html
表头说明: (result/03.circRNA_info/1.circRNA_annotation/*csv 鉴定的circRNA的注释信息表)
| 表头 | 说明 |
|---|---|
seqnames
|
染色体名称 |
start
|
circRNA的起始位置 |
end
|
circRNA的终止位置 |
width
|
circRNA长度 |
strand
|
circRNA位于参考序列的正链(+)或负链(-)上 |
source
|
注释来源,CIRIquant |
type
|
注释信息的类型,circRNA |
score
|
circRNA的CPM值 |
circ_id
|
circRNA名称 |
circ_type
|
circRNA类型,如exon / intron / intergenic / antisense |
bsj
|
反向拼接位点 (back-spliced junction) 的reads数量 |
fsj
|
可变剪切位点 (forward-spliced junction) 的reads数量 |
junc_ratio
|
环状NA与线性RNA的比值,计算方法为 2 * bsj / ( 2 * bsj + fsj) |
gene_id
|
host gene的ensemble id。'NA' 表示没有host gene,说明该circRNA的类型为 'intergenic' |
gene_name
|
host gene的HGNC symbol。'NA'同上 |
gene_type
|
host gene的类型。'NA'同上 |
2.2.2. 参考基因组比对
测序片段(fragments)是随机打断的,为了确定这些一段由哪些基因转录来,需要将质控后的clean reads比对到参考基因组上。使用HISAT2软件将Clean Reads与参考基因组进行快速精确的比对,获取Reads在参考基因组上的定位信息[4]。HISAT2软件官方手册。
如果参考基因组组装的较为完善,而且所测物种与参考基因组一致,且相关实验不存在污染,那么实验所产生的测序reads成功比对到基因组的比例会高于70% (Total Mapped Reads or Fragments)。本项目所用参考基因组为 hg38。
2.2.3. circRNA 预测及鉴定
使用CIRIquant鉴定 circRNA ,并预测 circRNA 的表达。目前发现的circRNAs主要来源于基因外显子exon,但还有其他类型,比如来源于内含子intron、基因间intergenic、反义链antisense。为了更进一步了解鉴定得到的circRNA详细信息,随后进行circRNA类型,circRNA 的长度分布,circRNA 的染色体分布分别进行分析,统计分析图如下。
|
result/03.circRNA_info/2.circRNA_length/Demo-input.png 
|
result/03.circRNA_info/2.circRNA_length/Demo-target.png 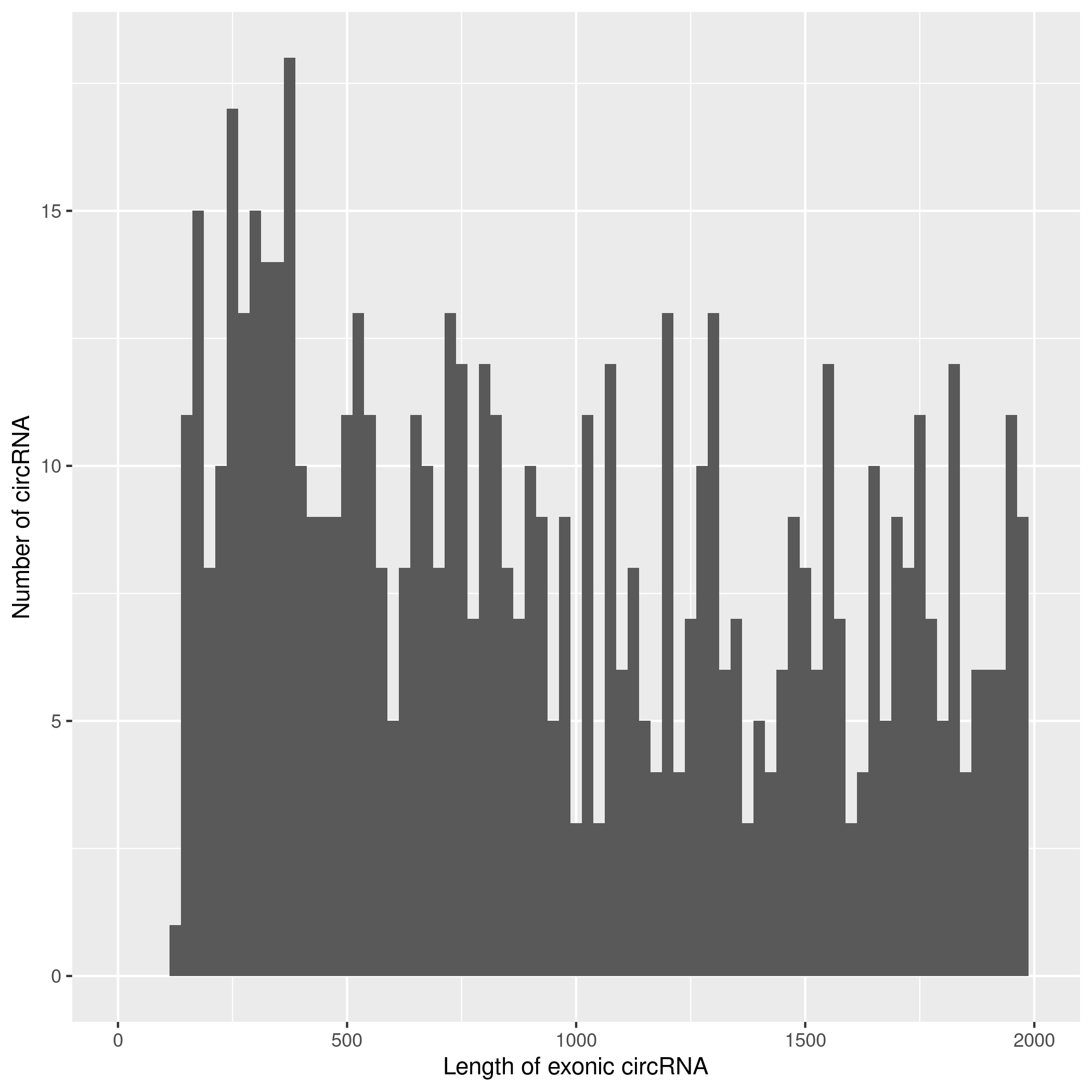
|
|
result/03.circRNA_info/3.circRNA_karyotype/Demo-input.png 
|
result/03.circRNA_info/3.circRNA_karyotype/Demo-target.png 
|
|
result/03.circRNA_info/4.circRNA_type/Demo-input_barplot.png 
|
result/03.circRNA_info/4.circRNA_type/Demo-target_barplot.png 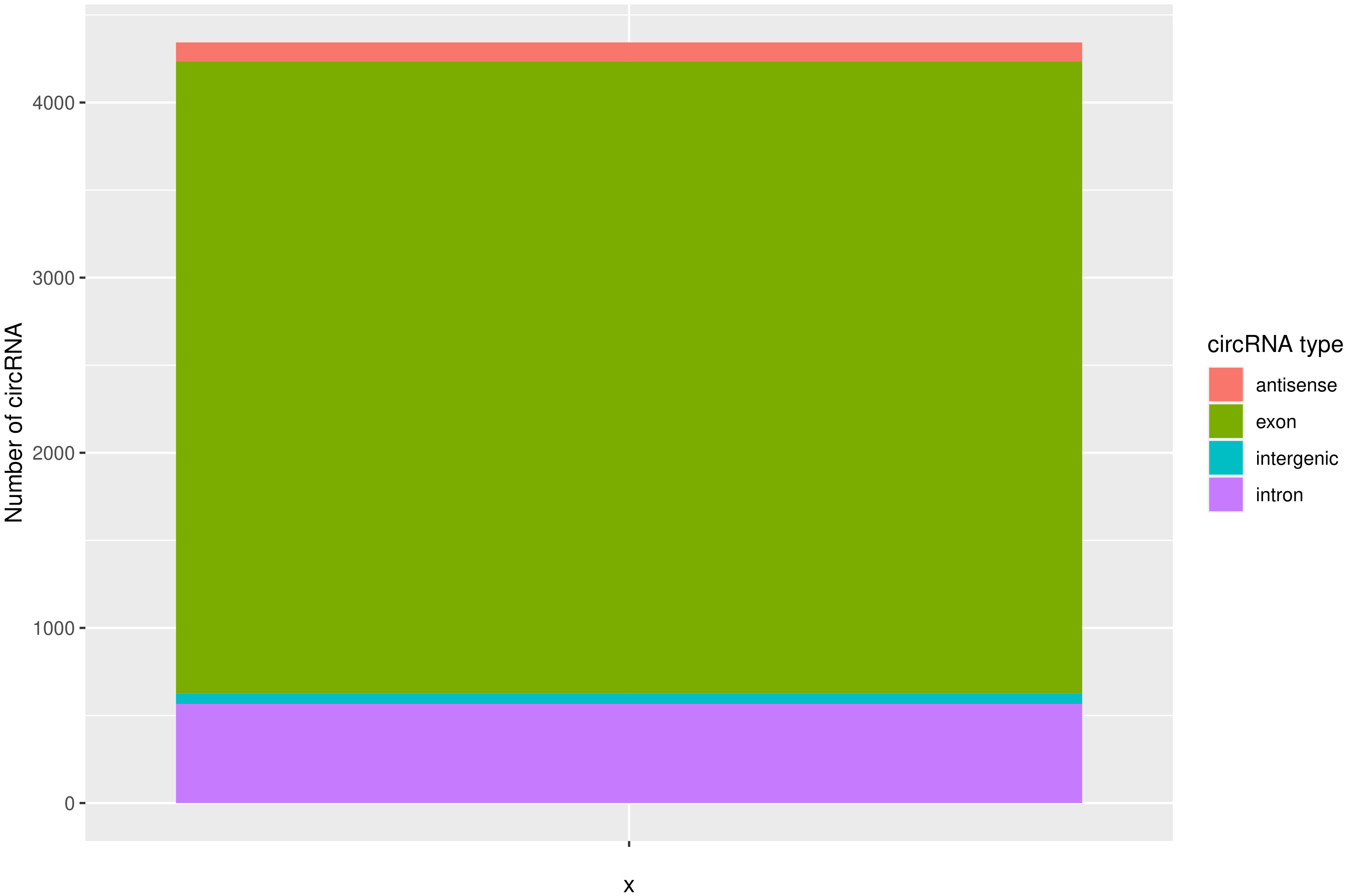
|
|
result/03.circRNA_info/4.circRNA_type/Demo-input_pie.png 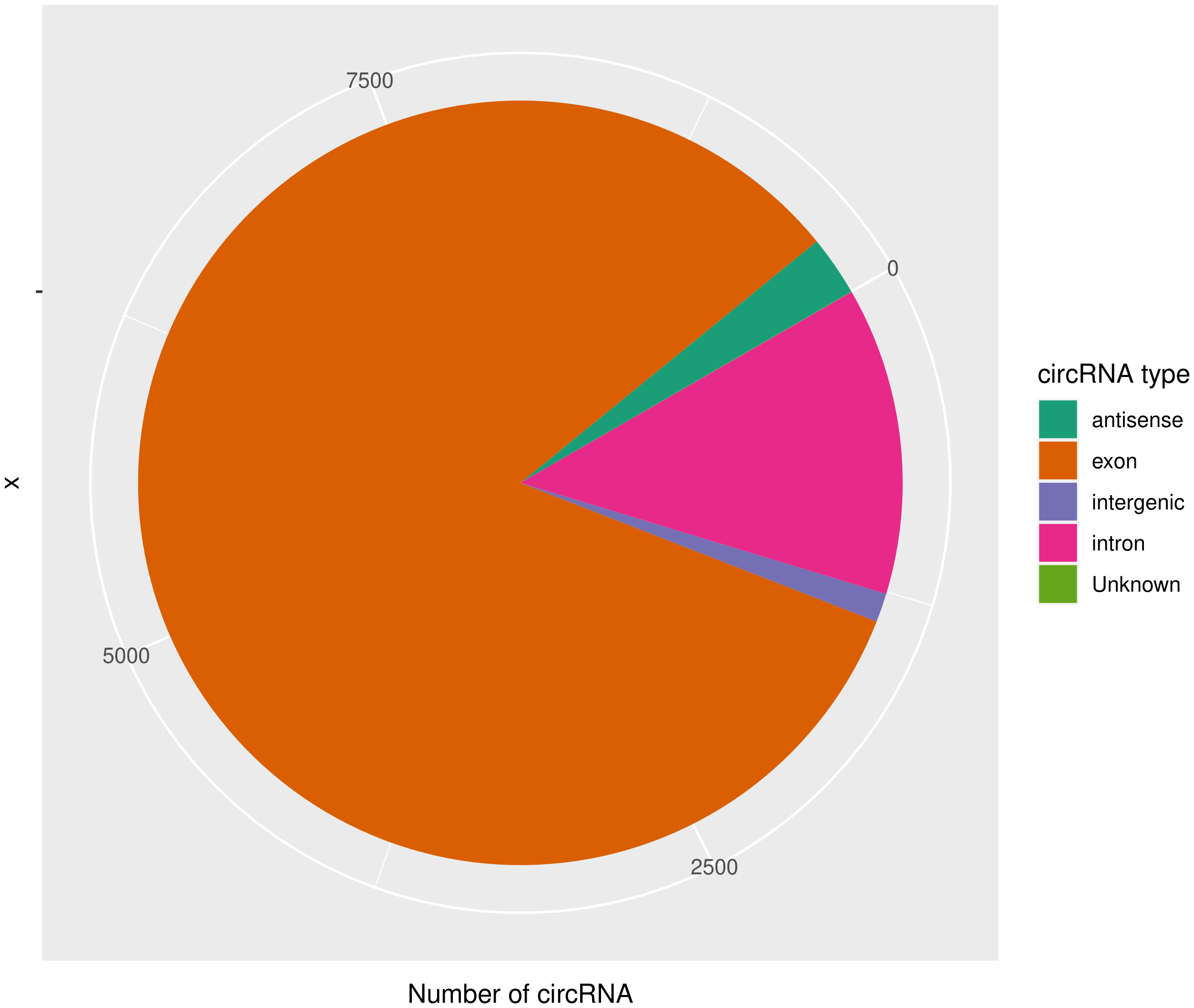
|
result/03.circRNA_info/4.circRNA_type/Demo-target_pie.png 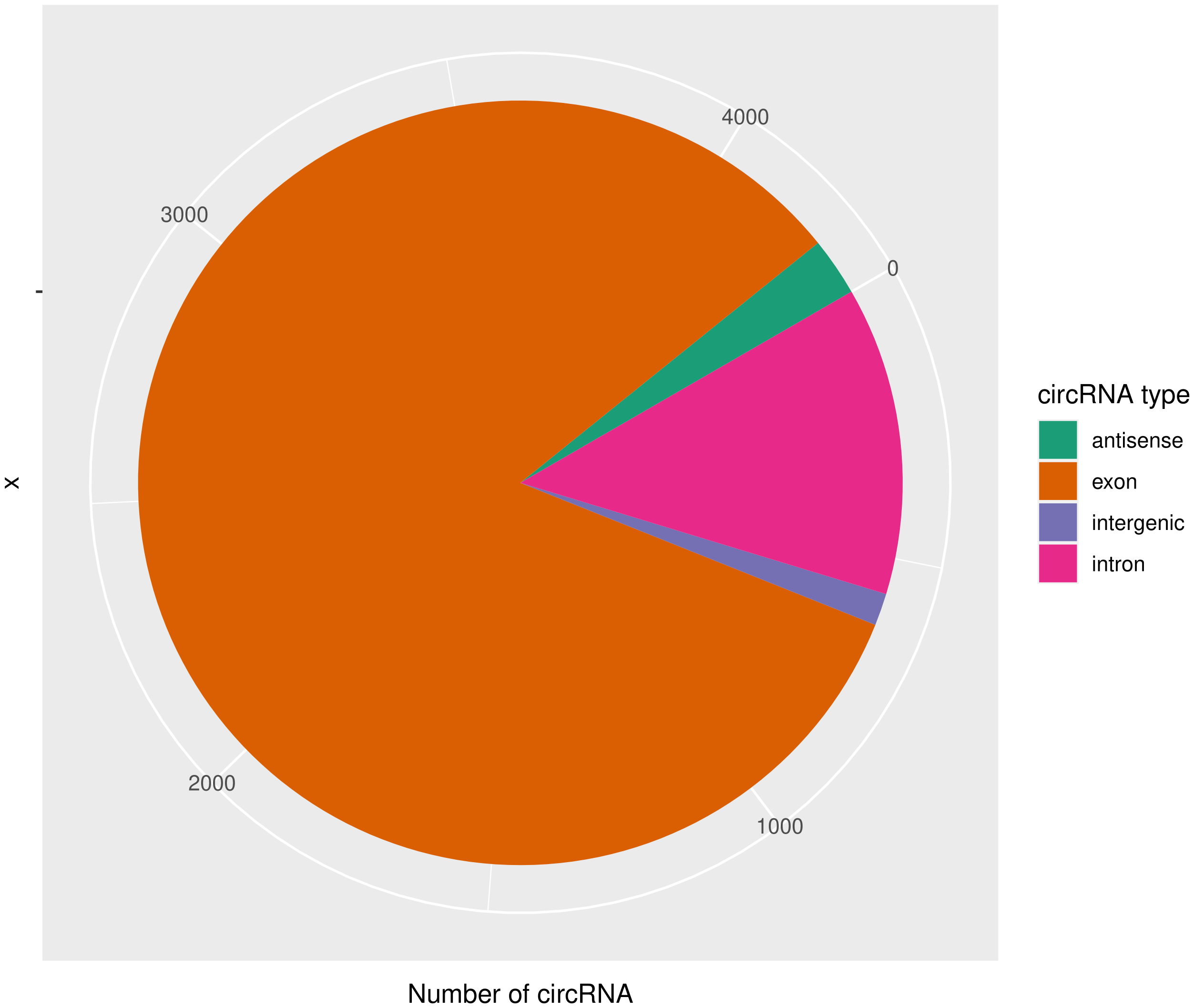
|
2.3. circRNA 差异分析
对于无重复样本,使用CIRIquant的CIRI_DE工具鉴定差异表达的circRNA。输出的 DE_score 综合了倍数变化和p值,从而提供了一种有效的方法来对差异表达的circRNA排名。此处我们筛选 |DE_score| > 1 作为显著差异表达结果。
2.3.1. 差异分析结果文件
| 文件路径 | 说明 |
|---|---|
result/04.DE/targetVSinput_deg_ALL.xls
|
circRNA差异分析结果(所有结果) |
result/04.DE/targetVSinput_deg_all-diff.xls
|
circRNA差异分析结果(筛选 |DE_score| > 1后)
|
result/04.DE/targetVSinput_deg_all-diff.bed
|
circRNA差异分析结果的bed文件(筛选 |DE_score| > 1后)
|
result/04.DE/targetVSinput_deg_up.xls
|
仅上调circRNA差异分析结果(筛选 DE_score > 1后)
|
result/04.DE/targetVSinput_deg_down.xls
|
仅下调circRNA差异分析结果(筛选 DE_score < -1后)
|
result/04.DE/diff-gene-types_count.txt
|
所有差异circRNA的host gene的类型统计 |
以上结果位于文件夹:result/04.DE/
表头说明: (result/04.DE/targetVSinput_deg*.xls差异分析结果文件)
| 表头 | 说明 |
|---|---|
circRNA_ID
|
circRNA名称 |
Case_BSJ
|
Case组 反向拼接位点 (back-spliced junction) 的reads数量 |
Case_FSJ
|
Case组 可变剪切位点 (forward-spliced junction) 的reads数量 |
Case_Ratio
|
Case组 环状NA与线性RNA的比值,计算方法为 2 * bsj / ( 2 * bsj + fsj) |
Ctrl_BSJ
|
Ctrl组 反向拼接位点 (back-spliced junction) 的reads数量 |
Ctrl_FSJ
|
Ctrl组 可变剪切位点 (forward-spliced junction) 的reads数量 |
Ctrl_Ratio
|
Ctrl组 环状NA与线性RNA的比值,计算方法为 2 * bsj / ( 2 * bsj + fsj) |
DE_score
|
差异表达分数(differential expression score) |
DS_score
|
差异剪切分数(differential splicing score) |
change
|
标注信息,'UP'表示显著上调,'DOWN'表示显著下调,'NOT'为表达没有显著变化。 |
circ_type
|
同上。circRNA类型,如exon / intron / intergenic / antisense |
gene_id
|
同上。host gene的ensemble id。'NA' 表示没有host gene,说明该circRNA的类型为 'intergenic' |
gene_name
|
同上。host gene的HGNC symbol。'NA'同上 |
gene_type
|
同上。host gene的类型。'NA'同上 |
2.3.2. 差异circRNA的基因组可视化
可将比对结果bw文件、CIRIquant鉴定得到的circRNA的bed文件、以及差异circRNA分析结果同时放入IGV查看,如:
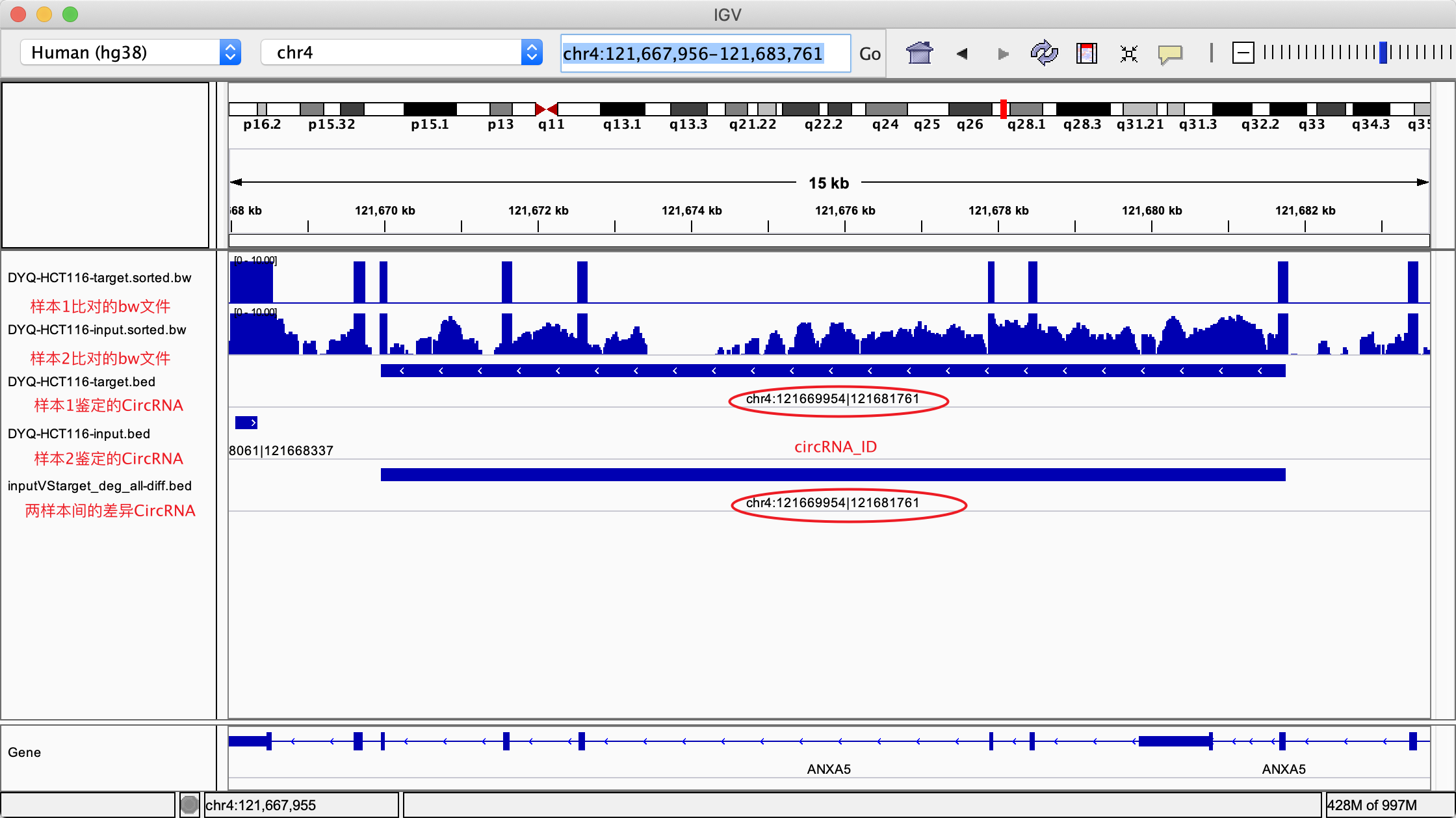
2.4. 差异circRNA宿主基因富集分析
我们将差异circRNA的宿主基因,挑选出仅为 protein coding 的基因,用这些基因进行后续富集分析。
我们根据基因表达量分析得到差异基因之后,必须进一步落到基因的功能上来。对于差异分析而言,往往涉及到成千上万个基因,这会使分析变得很复杂。解决思路是将一个基因列表分成多个部分,从而减少分析的复杂度。为了解决怎么分成不同类,通常会对基因功能进行富集分析, 期望发现在生物学过程中起关键作用的生物通路, 从而揭示和理解生物学过程的基本分子机制。功能富集分析可以将成百上千个基因、蛋白或者其他分子分到不同的通路中,以减少分析的复杂度。另外,在两种不同实验条件下,激活的通路显然比简单的基因或蛋白列表更有说服力。基因功能富集分析首先要构建基因集( gene set,如 GO 和 KEGG 数据库等),也就是基因组注释信息进行分类。然后再把我们的目标基因集(差异基因集或者其他基因集)映射到背景基因集上,注意区分注释与富集。
我们采用 clusterProfiler 软件对差异基因集进行 GO 功能富集分析, KEGG 通路富集分析等。富集分析基于超几何分布原理,其中差异基因集为差异显著分析所得差异基因并注释到 GO 或 KEGG 数据库的基因集,背景基因集为所有进行差异显著分析的基因并注释到 GO 或 KEGG 数据库的基因集。富集分析结果是对每个差异比较组合的所有差异基因集、上调差异基因集、下调差异基因集进行富集。本报告中展示的表格是选取某一个比较组合的富集分析结果,图片是部分富集分析结果。
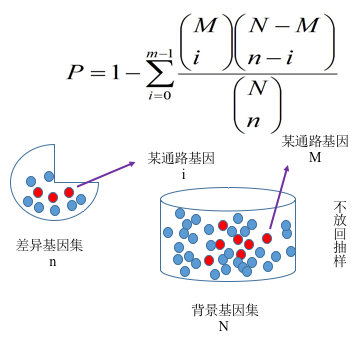
图 5 基因富集分析原理图
2.4.1. 富集分析结果文件
| 结果路径 | 结果说明 |
|---|---|
| GO富集分析结果 |
|
result/05.Enrichment/*/gene.ego_all-p.adjust1.00.csv
|
GO富集结果列表(所有结果) |
result/05.Enrichment/*/gene.ego_all-p.adjust0.05.csv
|
GO富集结果列表(按p.adj<0.05筛选后) |
result/05.Enrichment/*/gene.ego_ALL.csv
|
GO富集结果列表(MF、BP、CC所有结果) |
result/05.Enrichment/*/gene.GO-*-barplot.p*
|
GO富集分析柱状图 |
result/05.Enrichment/*/gene.GO-*-dotplot.p*
|
GO富集分析散点图 |
result/05.Enrichment/*/gene.GO-*-DAG.p*
|
GO富集分析DAG图 |
| KEGG富集分析结果 |
|
result/05.Enrichment/*/gene.KEGG.csv
|
KEGG富集结果列表(所有) |
result/05.Enrichment/*/gene.KEGG_significant.csv
|
KEGG富集结果列表(按p.adj<0.05筛选后) |
result/05.Enrichment/*/gene.KEGG-*-barplot.p*
|
KEGG富集分析柱状图 |
result/05.Enrichment/*/gene.KEGG-*-dotplot.p*
|
KEGG富集分析散点图 |
以上结果位于文件夹:result/05.Enrichment/
网页预览图:
表头说明: (result/05.Enrichment/*/gene.ego_*.csv GO富集结果列表)
| ID | 对应GO数据库中的ID |
|---|---|
| ONTOLOGY | 分子功能(Molecular Function),生物过程(biological process)和细胞组成(cellular component) |
| Description | GO的描述 |
| GeneRatio | 对应GO 差异基因数 / 能够对应到GO数据库中同类型的差异基因数 |
| BgRatio | 对应GO包含对应物种的基因数 / GO数据库中包含对应物种的基因数 |
| pvalue | 富集分析得到的p-value |
| p.adjust | 校正后的p-value |
| qvalue | 富集分析得到的qvalue |
| geneID | 富集基因列表 |
| Count | 富集基因数目 |
表头说明: (result/05.Enrichment/*/gene.KEGG*.csv KEGG富集结果列表)
| ID | 对应PATHWAY数据库中的ID |
|---|---|
| Description | PATHWAY的描述 |
| GeneRatio | 对应PATHWAY 差异基因数 / 能够对应到PATHWAY数据库中的差异基因数 |
| BgRatio | 对应PATHWAY包含对应物种的基因数 / PATHWAY数据库中包含对应物种的基因数 |
| pvalue | 富集分析得到的p-value |
| p.adjust | 校正后的p-value |
| qvalue | 富集分析得到的qvalue |
| geneID | 富集基因列表 |
| Count | 富集基因数目 |
2.4.2. GO功能富集分析
GO(Gene Ontology) 是描述基因功能的综合性数据库,可分为生物过程( biological process )和细胞组成( cellular component )分子功能( Molecular Function )三个部分。 GO 功能富集以 padj 小于 0.05 作为为显著性富集的阈值,富集结果见结果文件。
从 GO 富集分析结果中,选取最显著的 30 个 Term 绘制柱状图进行展示,若不足 30 个,则绘制所有 Term ,按生物过程、细胞组分和分子功能三大类别及差异基因上下调分类画的柱状图。
有向无环图 (Directed Acyclic Graph,DAG) 为差异基因 GO 富集分析结果的图形化展示方式。图中,分支代表包含关系,从上至下所定义的功能范围越来越小,选取每个差异比较组合的 GO 富集结果最显著性前 5 位的 GO Term 作为有向无环图的主节点,并通过包含关系,将相关联的 GO Term 一起展示,颜色的深浅代表富集程度。我们的项目中分别绘制生物过程、分子功能和细胞组分的 DAG 图。

图 6 GO富集分析柱状图
图中纵坐标为GO Term,横坐标为GO Term富集的显著性水平,数值越高越显著

图 7 GO富集分析散点图
图中横坐标为注释到GO Term上的差异基因数与差异基因总数的比值,纵坐标为GO Term
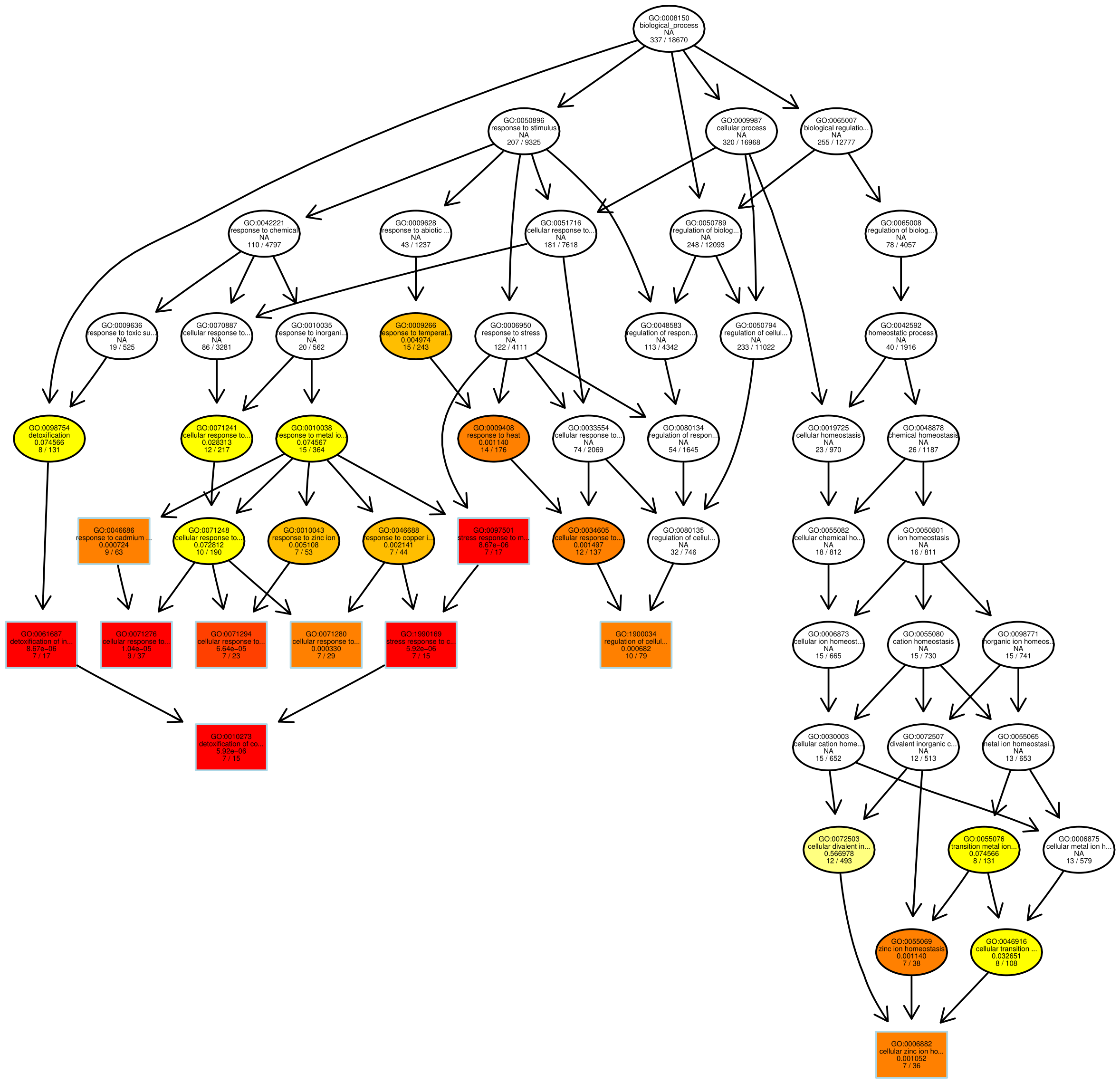
图 8 GO富集分析DAG图
每个节点代表一个GO术语,方框代表的是富集程度为TOP5的GO,颜色的深浅代表富集程度,颜色越深就表示富集程度越高,每个节点上展示了该TERM的名称及富集分析的padj
2.4.3. KEGG通路富集分析
KEGG(Kyoto Encyclopedia of Genes and Genomes) 是整合了基因组、化学和系统功能信息的综合性数据库。 KEGG 通路富集以 padj 小于 0.05 作为显著性富集的阈值,富集结果见结果文件。
从 KEGG 富集结果中,选取最显著的 20 个 KEGG 通路绘制柱状图进行展示,若不足 20 个,则绘制所有通路,如下图所示。图中横坐标为通路富集的显著性水平,数值越高越显著,纵坐标为 KEGG 通路。
从 KEGG 富集结果中,选取最显著的 20个KEGG 通路绘制散点图进行展示,若不足 20 个,则绘制所有通路,如下图所示。图中横坐标为注释到 KEGG 通路上的差异基因数与差异基因总数的比值,纵坐标为 KEGG 通路,点的大小代表注释到 KEGG 通路上的基因数,颜色从红到紫代表富集的显著性大小。
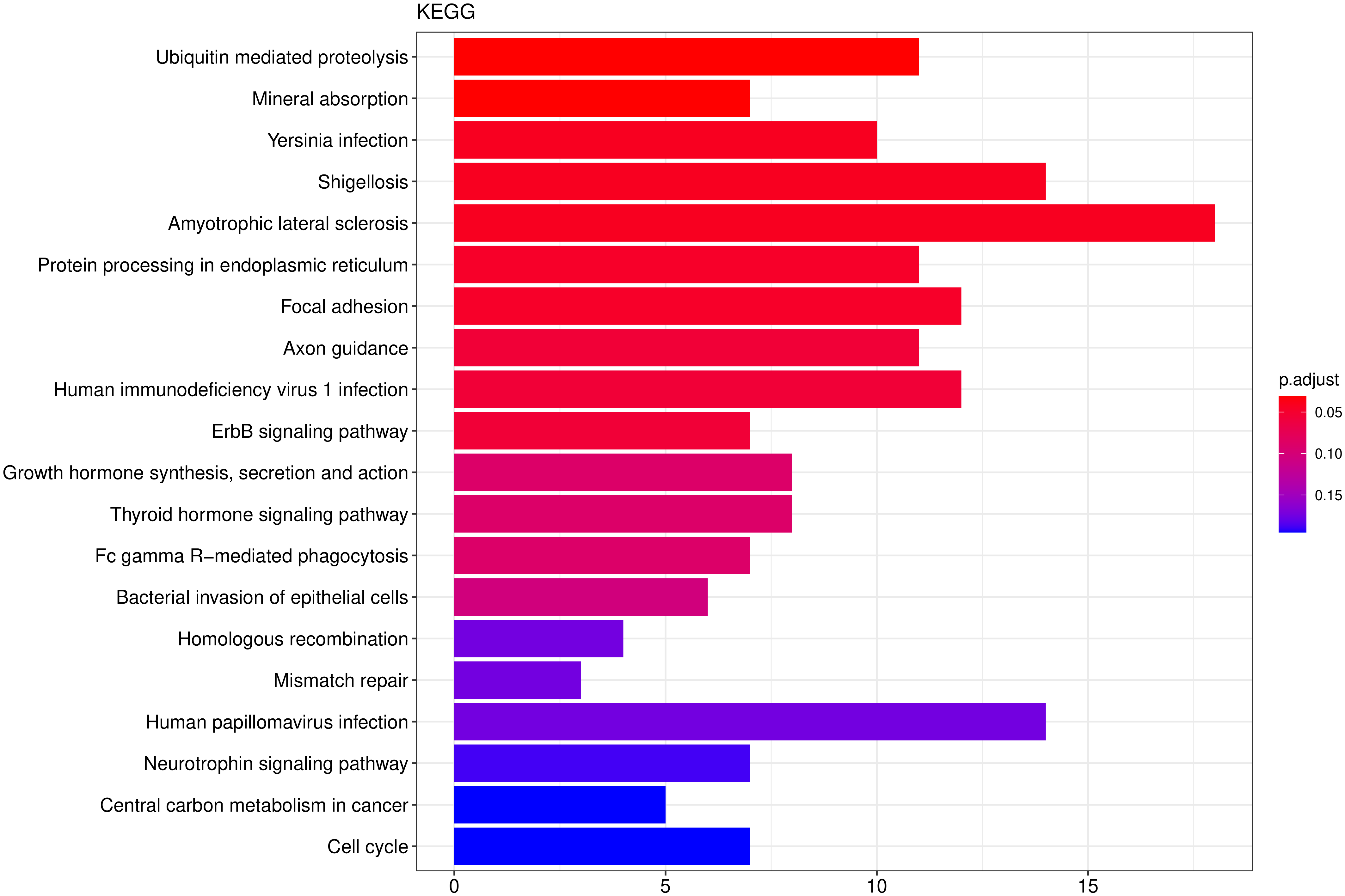
图 9 KEGG富集分析柱状图
图中横坐标为通路富集的显著性水平,数值越高越显著,纵坐标为KEGG通路。
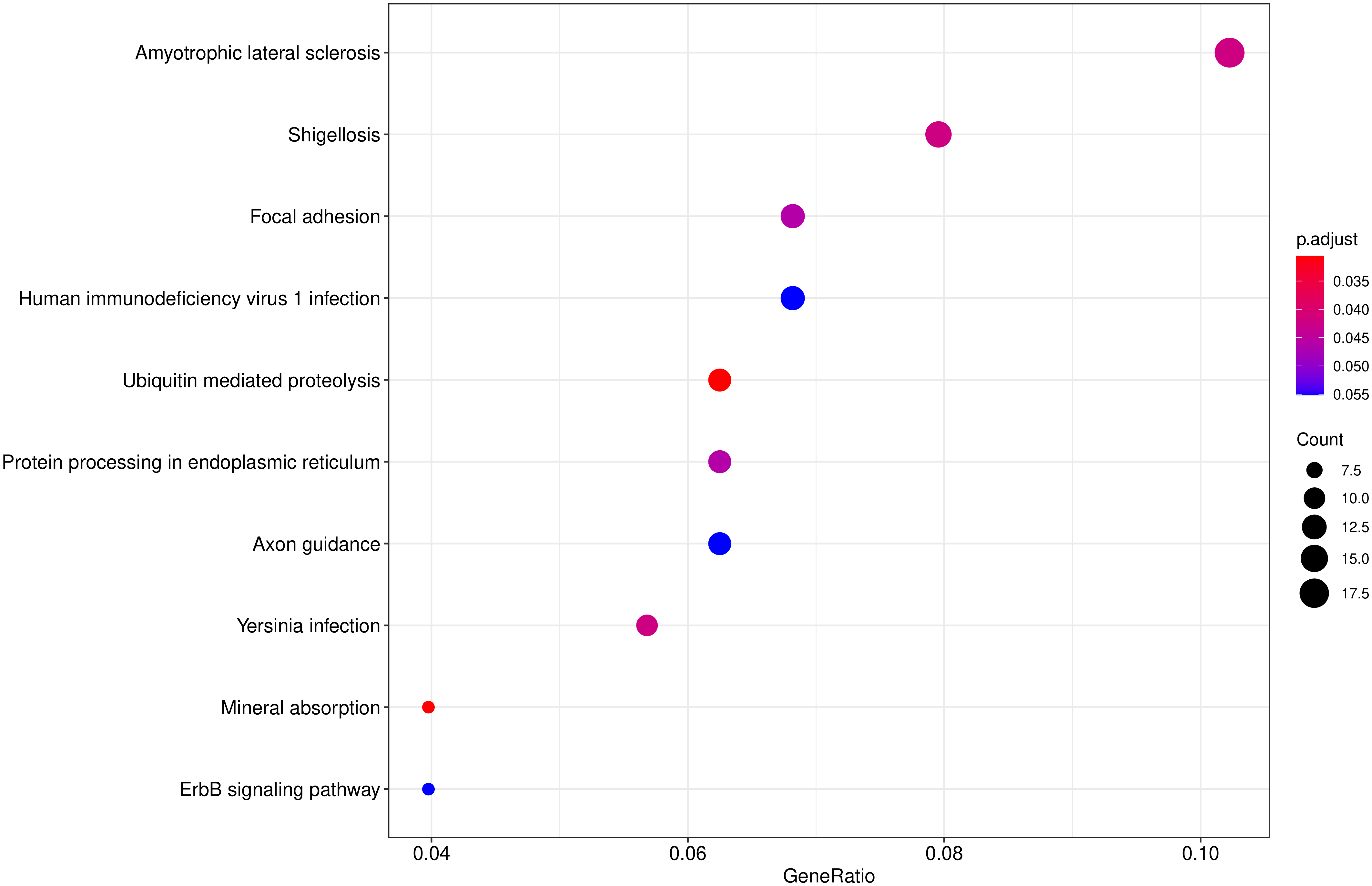
图 10 KEGG富集散点图
图中横坐标为注释到KEGG通路上的差异基因数与差异基因总数的比值,纵坐标为KEGG通路
- 本文固定链接: https://maimengkong.com/zu/1292.html
- 转载请注明: : 萌小白 2022年11月20日 于 卖萌控的博客 发表
- 百度已收录
