1. 工作流程
RNA免疫共沉淀(RIP)是一种用于研究蛋白质与 RNA 的体内相互作用的经典实验技术。采用特异性抗体将目的蛋白进行免疫沉淀,由此可以把目的蛋白所结合的RNA片段也富集下来。通过与高通量测序技术的结合,对 RIP 后的RNA 产物进行测序分析, 从全基因组范围内寻找目的蛋白的 RNA 结合位点,以高效率的测序手段得到高通量的数据结果。
1.1. RIP 免疫沉淀实验流程
目前主要有两种不同的RIP 实验方法,大致流程如下(以细胞样品的处理过程为例):
RNA Immunoprecipitation
-
准备足量的新鲜细胞,每个IP约1x107个细胞,用RIP裂解液裂解细胞
-
加入2-5ug抗体,抗体与蛋白,4℃孵育过夜
-
加入proteinA/G磁珠,4℃孵育4-6小时
-
清洗磁珠。
-
Proteinase K 解交连。
-
酚氯仿或RNA提取试剂盒提取RNA
-
QPCR 检测或建库测序
1.2. RIP Sequencing 文库构建流程
-
用qubit 及2100对RIP片段进行定量及片段长度检测
-
加入适当的Mg2+,加热打断RNA片段
-
加入反转录酶,反转录成cDNA
-
断裂RNA链且以断裂RNA为引物,cDNA为模版,形成双链DNA
-
补齐片段末端,并在3’末端加A尾
-
添加Adapter
-
0.8X AMPure beads去掉多余的Adapter
-
文库PCR扩增
-
1XAMPure beads 去掉多余的primer
-
qPCR测定文库浓度
-
Agilent 2100测定文库片段大小
1.3. 生物信息分析流程
将测序结果与参考基因组比对,比对上唯一位置的序列用于后续标准信息分析及个性化分析。信息分析流程如下:
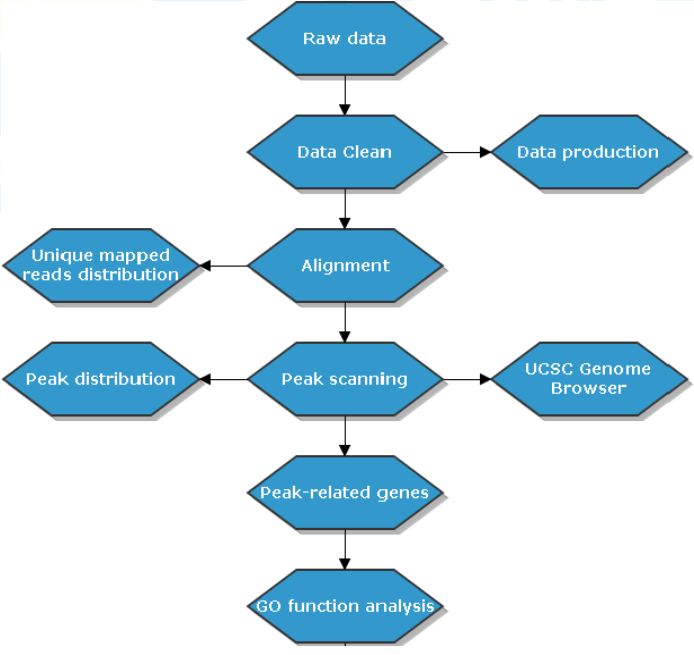
2. 生物信息分析
2.1. RIP Sequencing 文库质检结果
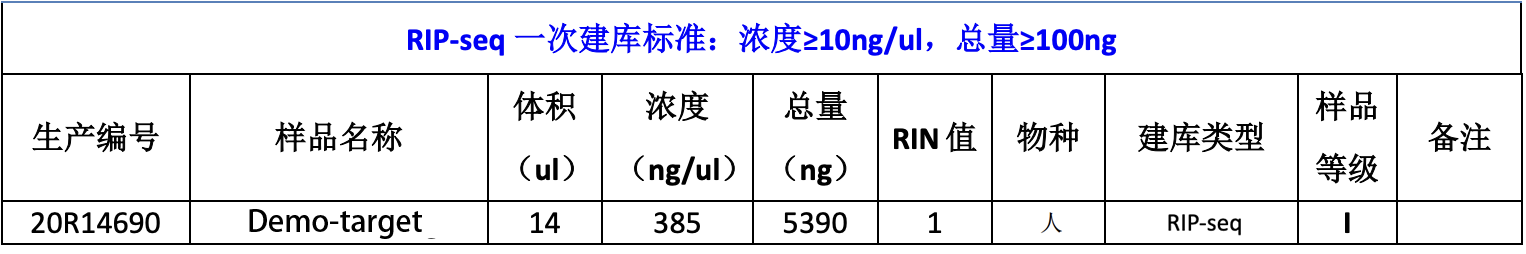
文库片段质检,RIP文库的染色质片段在150-300bp之间,建库加入约140bp的接头后,片段应该分布在300-450bp之间为最好。

Ladder 自下而上依次为 25(绿色),200,500,1000,2000,4000nt
2.2. 测序数据质量控制
对原始测序数据及去除接头后的可用数据进行质量评估。RIP数据一般为双端测序,因此,每个测序样本会有两个测序结果。
评估的具体内容见:
| 结果说明 | 结果路径 |
|---|---|
| RawData-fastqc 文件链接 | /Results/01.qc/qc_rawdata/*.html |
| CleanData-fastqc 文件链接 | /Results/01.qc/qc_cleandata/*.html |
| Fastqc 格式补充说明 | /Results/01.qc/qc_Supplement/qc_Supplement.html |
以上结果均位于文件夹:/Results/01.qc/
2.3. Peak calling数据统计结果
质检后的reads,采用trim-galory对reads进行去接头,去接头后,再次对reads进行质检,主要检测接头是否去除干净。去除接头的reads,用hisat2软件将reads mapping到基因组上,得到reads在基因组上的信息,即.bam文件,将input的.bam文件与IP的.bam文件,通过MASC2进行 peak calling,得到peak文件,即为.bed文件,对得到的peak进行注释,并进行功能分析。
采用常用 reads 富集峰鉴定软件 MACS2 在全基因范围进行 peak 扫描,得到 Peak 在基因组上的位置信息、peak 富集信息等。
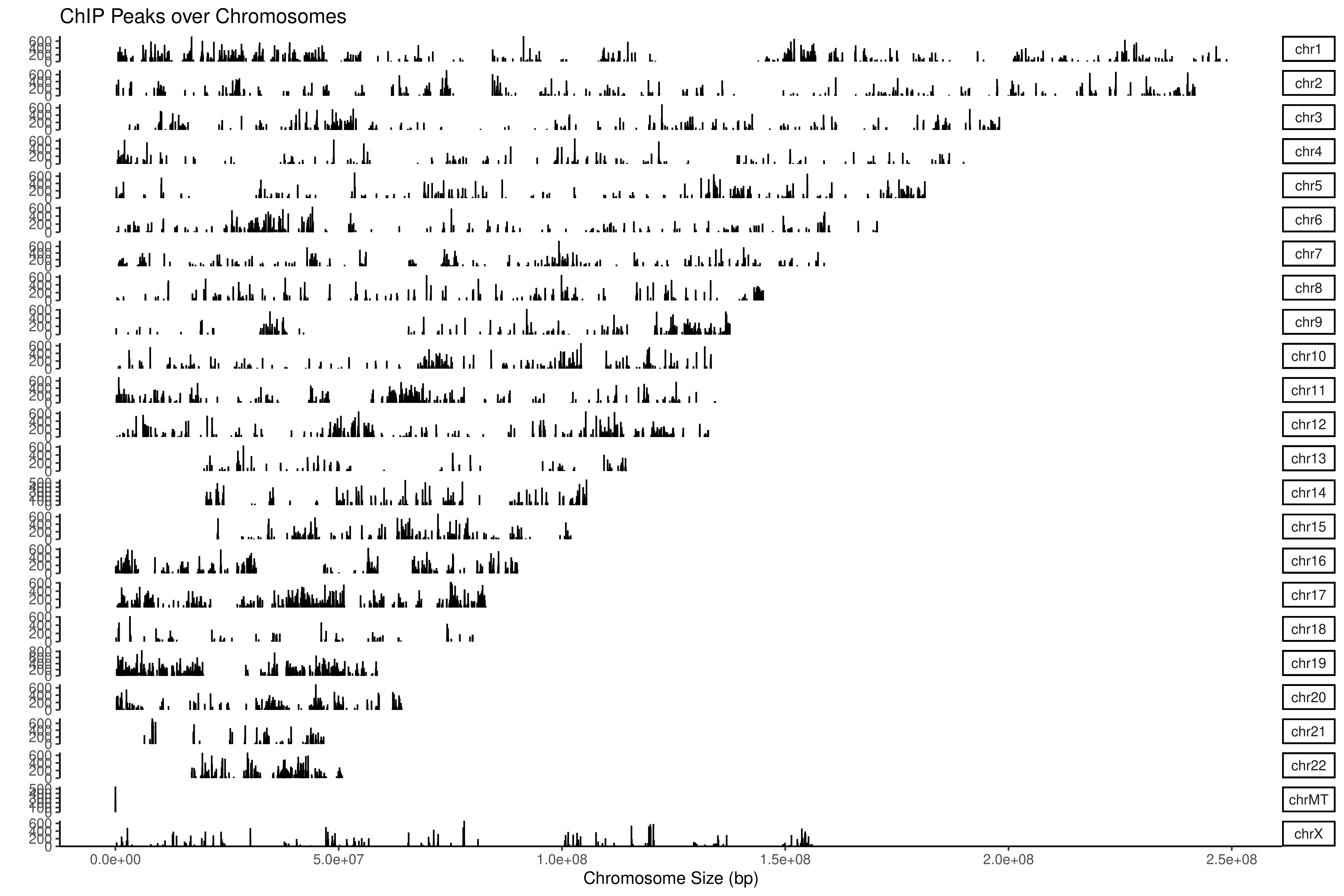
图 2.4.1 全基因组 Reads 富集峰
使用Chipseeker对Reads富集峰进行注释,得到tss上下游3k的基因注释信息。使用bedtools对富集峰与lncRNA取交集得到所在基因,将得到的lncRNA基因同样使用chipseeker(仅用 Protein-coding 注释)进行上下游10k的临近基因注释,对基因结果进行后续富集分析。
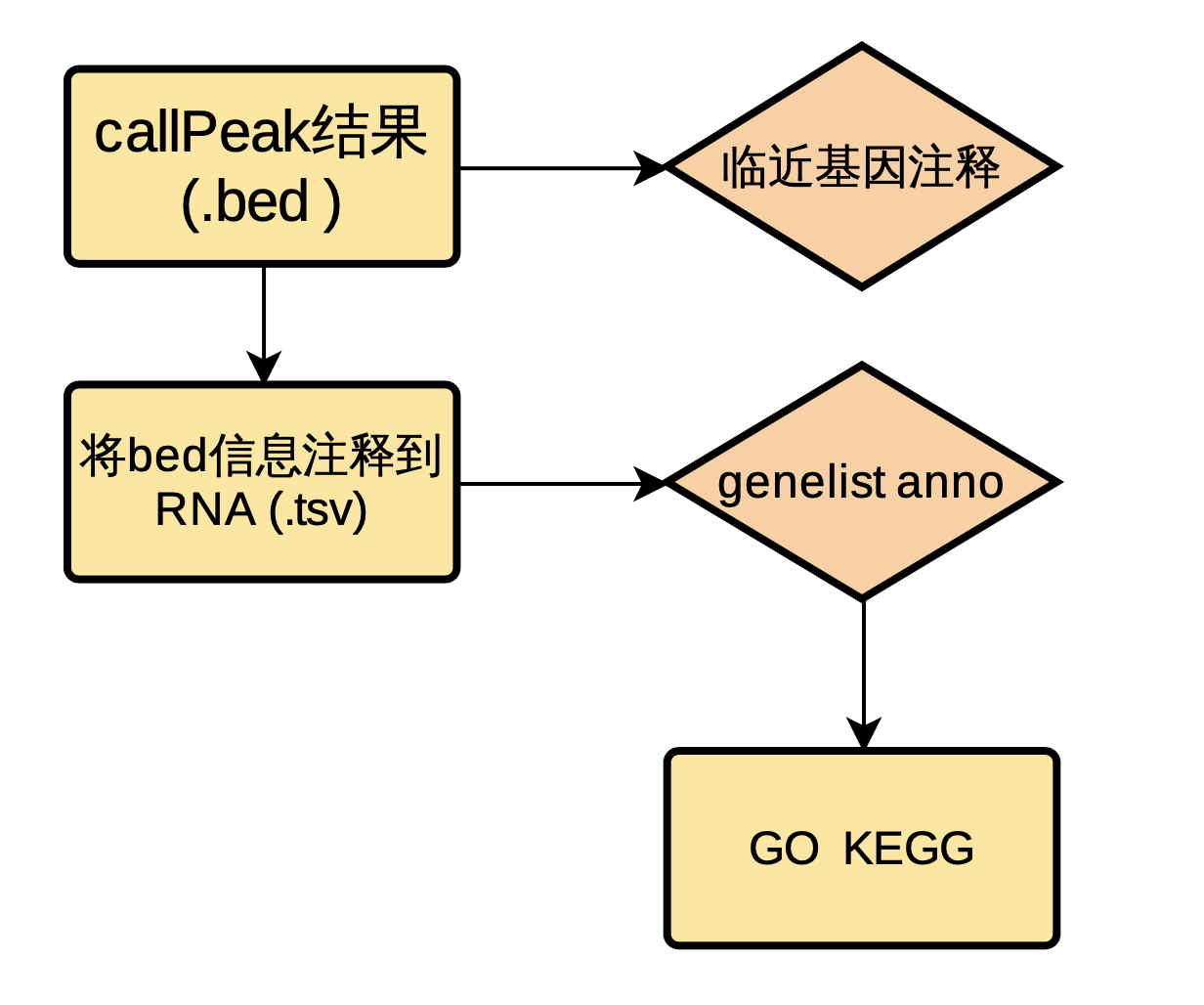
图 2.4.2 Peak信息anno流程图
结果文件:
| 结果说明 | 结果路径 |
|---|---|
| reads在基因组上的分布信息 |
DYQ-HCT116-target.bw
|
| callPeak peak信息 |
DYQ-HCT116_peaks.bed
|
| callPeak tss上下游3k注释信息 |
DYQ-HCT116_peaks.PeakAnno.xls
|
| callPeak 与 lncRNA 交集peak信息 |
DYQ-HCT116_peaks_lncRNA.bed
|
| 交集peak信息与临近Protein-coding的注释信息 |
Peak_LncRNA_Anno_Protein-coding.xls
|
| 交集peak信息与临近Protein-coding的注释信息(bed) |
Peak_LncRNA_Anno_Protein-coding.bed
|
| 注释信息的基因信息汇总 |
gene.list.annoinfo.xls
|
以上结果均位于文件夹: /Results/02.callPeak
2.4. Peak 基因注释与 GO 功能分析
Peak 所在基因进行GO 功能分析,并按照基因功能进行聚类分析。y轴为基因的功能聚类,x轴为基因count数,颜色为校正p值。GO功能富集以padj小于0.05作为为显著性富集的阈值,GO分析有3种类型,分别为CC(细胞组分),MF(分子功能),BP(生物过程)。富集结果见:
条形图纵坐标为GO Term,纵坐标为count数,颜色从红到紫代表富集的显著性大小。
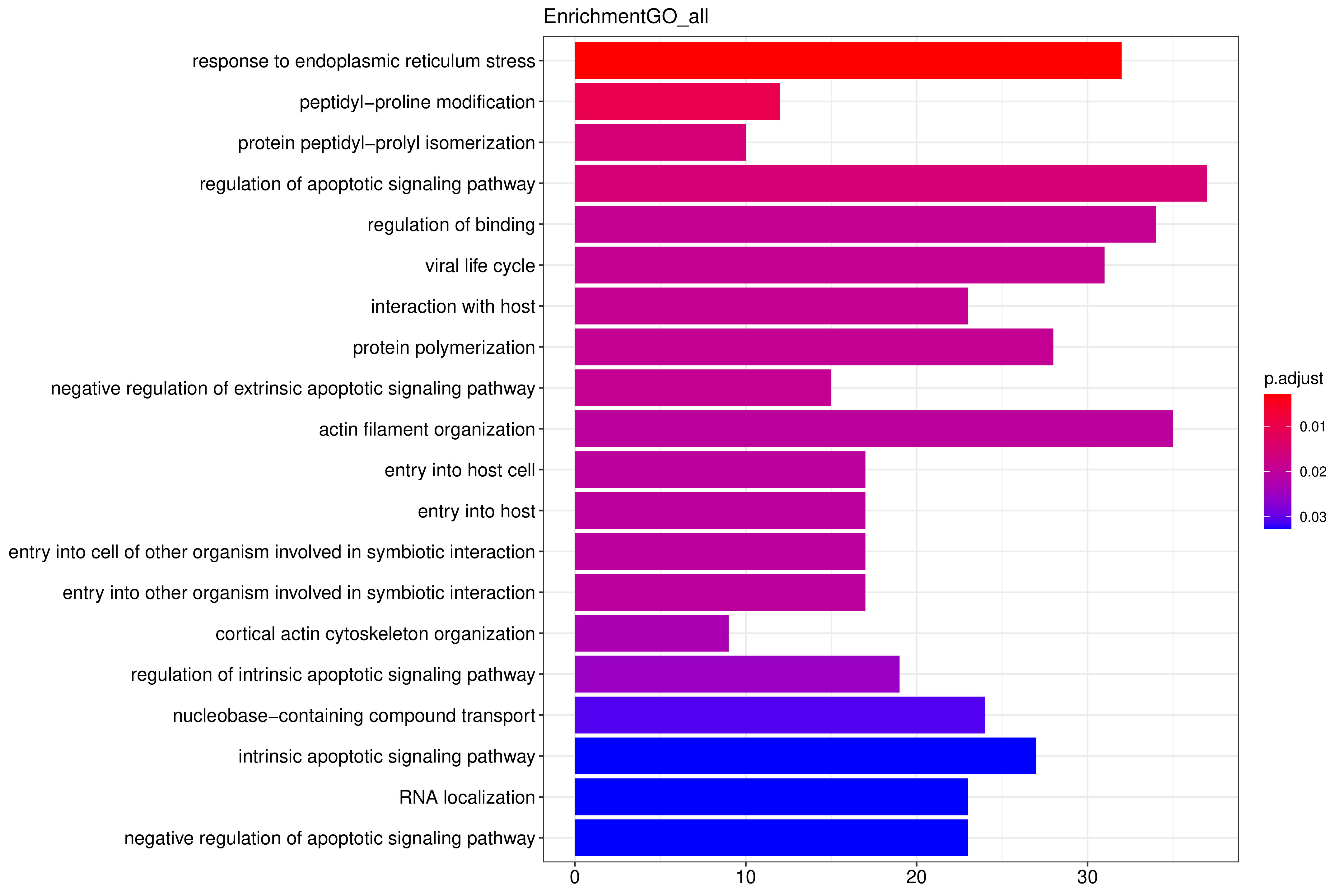
图 2.5.1 Peak 相关基因的GO 功能富集分析(条形图)
气泡图纵坐标为GO Term,点的大小代表注释到GO Term上的基因数,颜色从红到紫代表富集的显著性大小
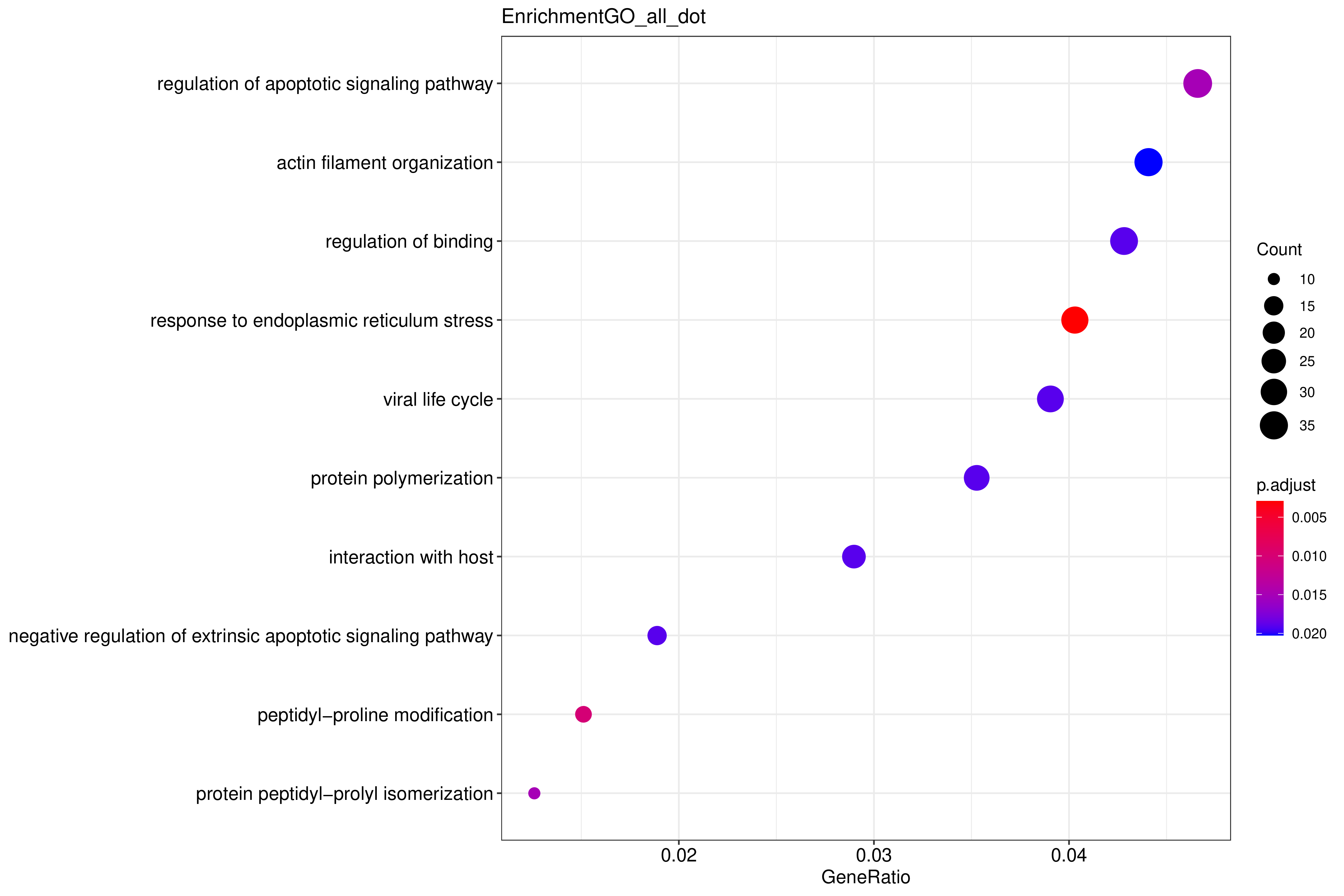
图 2.5.2 Peak 相关基因的 GO 功能富集分析(气泡图)
结果文件:
2.5. Peak 基因注释与 KEGG通路分析
KEGG(Kyoto Encyclopedia of Genes and Genomes)是整合了基因组、化学和系统功能信息的综合性数据库。KEGG通路富集以padj小于0.05作为显著性富集的阈值,富集结果如下表所示,见结果文件:Enrichment/KEGG。 从KEGG富集结果中,选取最显著的20个KEGG通路绘制柱状图进行展示,若不足20个,则绘制所有通路,如下图所示。图中横坐标为KEGG通路,纵坐标为通路富集的显著性水平,数值越高越显著。
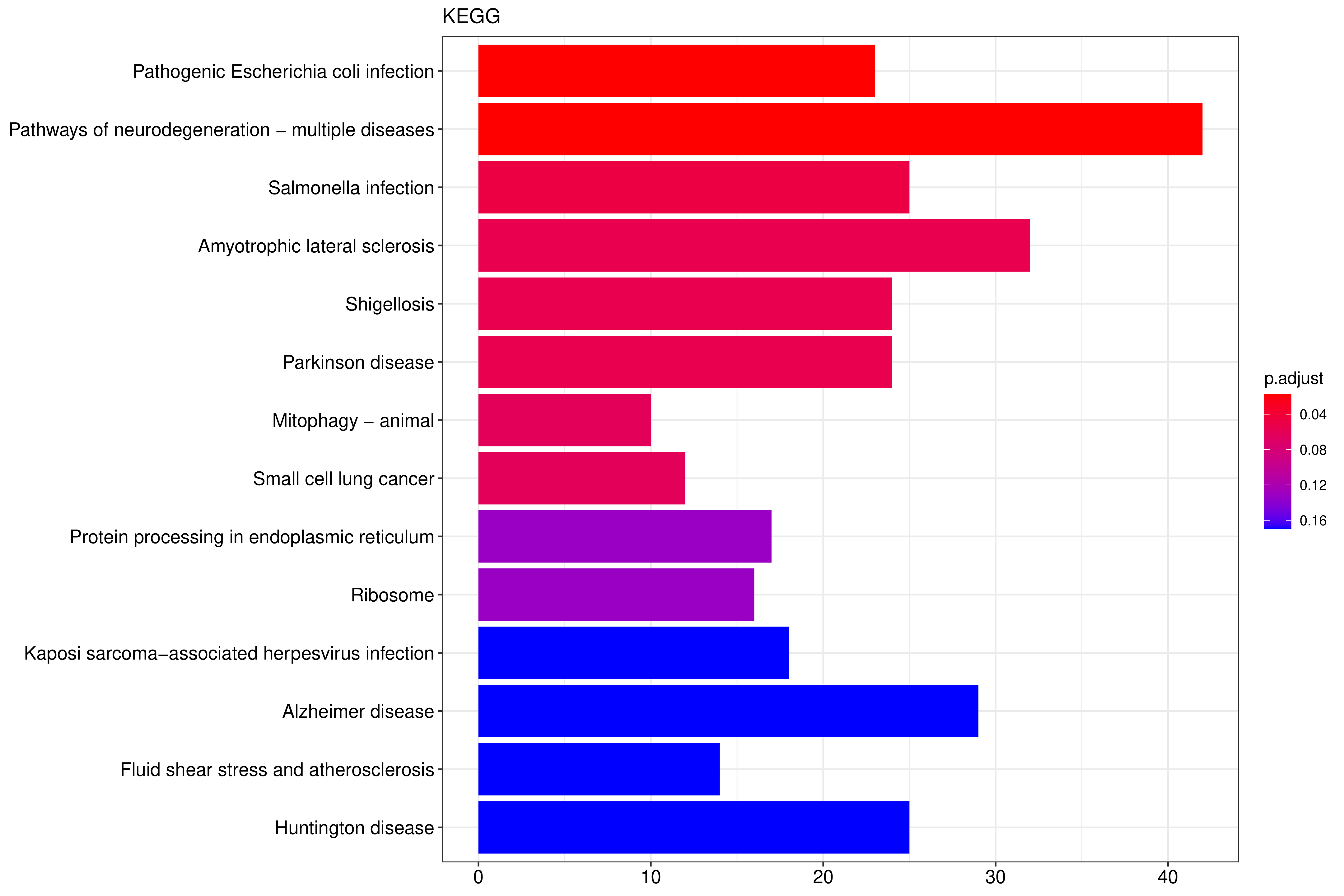
图 2.6.1 Peak 相关基因的KEGG通路富集分析(条形图)
气泡图纵坐标为GO Term,点的大小代表注释到GO Term上的基因数,颜色从红到紫代表富集的显著性大小
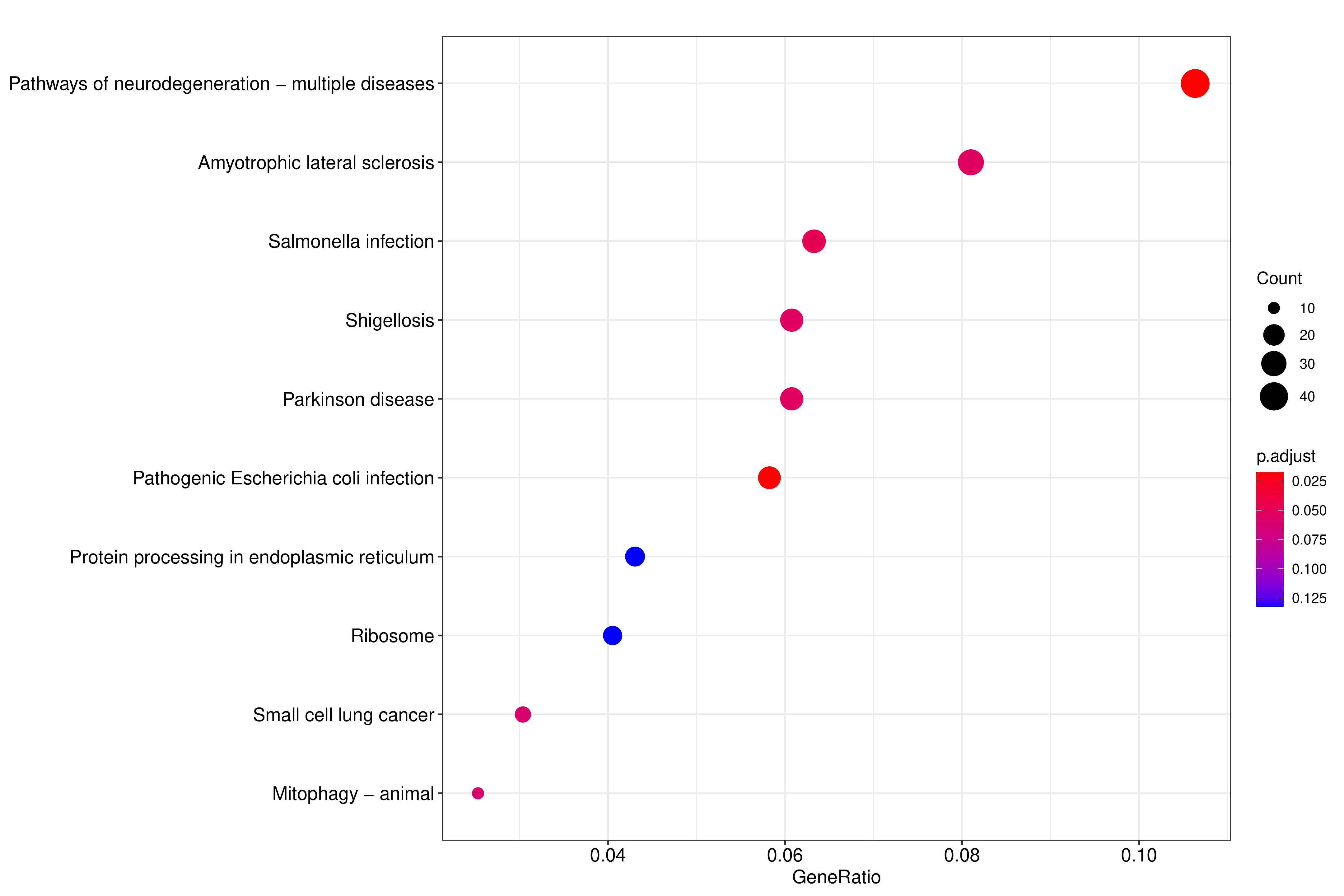
图 2.6.2 Peak 相关基因的KEGG通路富集分析(气泡图)
结果文件:
- 本文固定链接: https://maimengkong.com/zu/1291.html
- 转载请注明: : 萌小白 2022年11月20日 于 卖萌控的博客 发表
- 百度已收录
