全基因组关联分析(GWAS, Genome-wide association studies)是研究复杂疾病的遗传基础的策略之一。在肿瘤研究领域中,几乎所有常见恶性肿瘤的GWAS均已完成,并确定了与风险增加相关的450多个遗传变异。这些研究不仅揭示了致癌的新途径,而且还表明了常见的遗传变异大幅增加了许多常见癌症的遗传风险。GWAS有望应用于药物研发和癌症预防,助力精准预测、诊断和治疗。当前GWAS面临将SNP转换成复杂疾病的致病分子机制的挑战,需要破译其关联的功能和生物学基础,这在一定程度上阻碍了GWAS的应用。
 图片来自:scienceblog
图片来自:scienceblog
一、GWAS的出现
癌症全基因组关联分析发现了两类主要的癌症易感性变异,它们在一般人群中具有不同程度的风险和患病率。第一,经直接评估候选基因获得的罕见中外显率变异(风险等位基因频率<2%;ORs>2.0)。例如,除BRCA1和BRCA2之外,其他编码DNA损伤反应通路相关蛋白的基因,如ATM、CHEK2以及PALB2都与乳腺癌风险增加相关。由于它们的风险等位基因频率在0.1%到0.5%之间,并且每一种基因的风险仅增加了两倍,这些基因的变异对乳腺癌家族风险的贡献很小。第二,经GWAS鉴定获得的常见低外显率等位基因(风险等位基因频率>5%;ORs<1.5)。然而,许多癌症的外显率谱和风险等位基因的频率可能是连续的。这种二分法可能仅反映了最容易被发现的风险等位基因的子群,而不是潜在的生物或进化约束。
对常见遗传变异的深入研究和高通量基因分型技术的发展,使GWAS成为可能。GWAS通过一种基于基因组的方法,比较了大量不相关的癌症患者和匹配的健康个体的常见DNA变异的频率,从而确定与癌症风险相关的遗传变异。当前大多数常见癌症的GWAS已经完成,与其风险相关的遗传变异已得到确定,这为多基因易感性提供了直接证据。
二、GWAS的研究设计
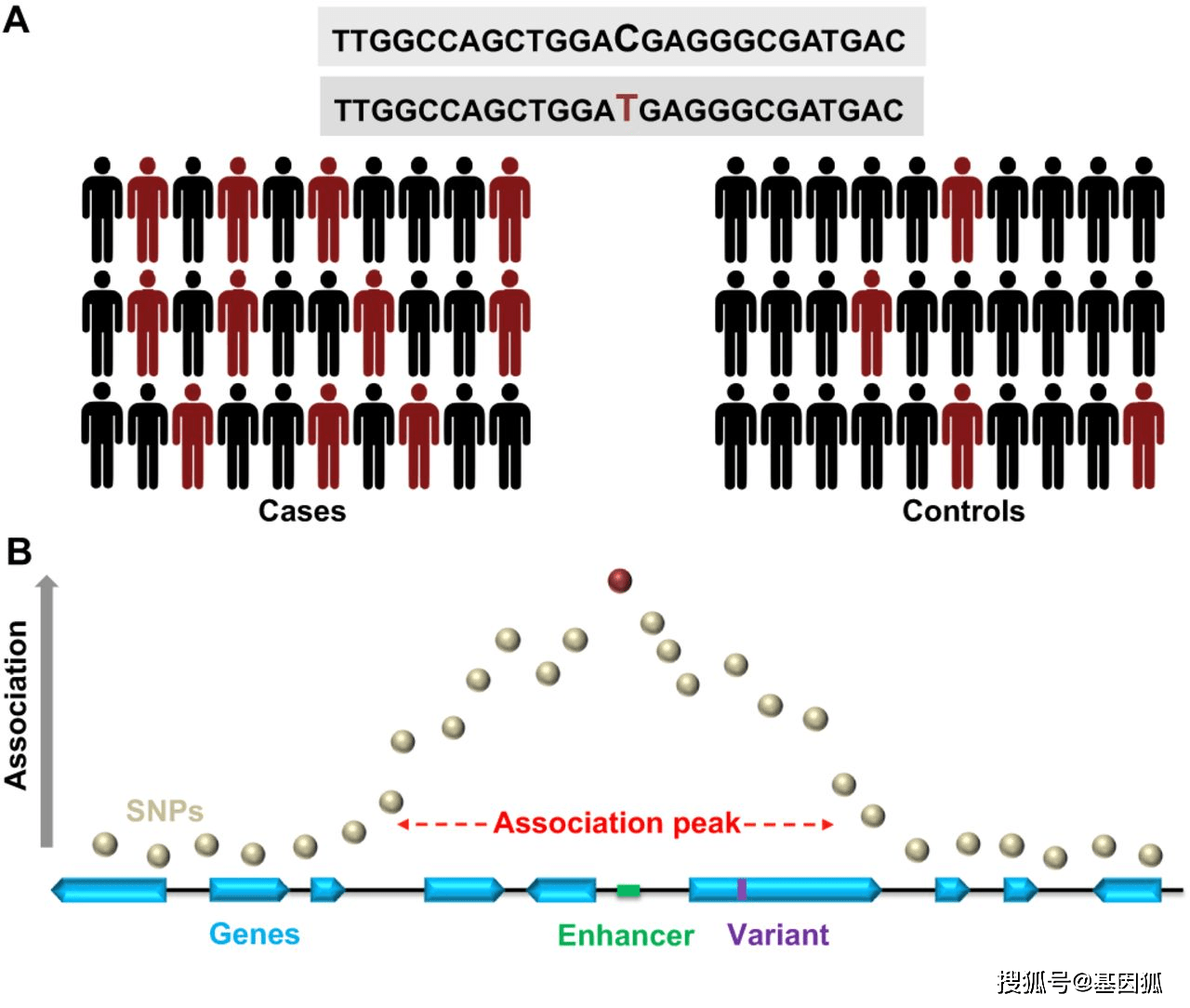
GWAS基于相邻的DNA片段往往是非独立共同遗传的原理,借助tag SNP在高度连锁不平衡(LD)的基因组区域中的代表性,从而减少需要分型的SNP数量。通过检测哪些SNP等位基因在患者中出现的频率高于或低于对照组,从而确定与风险相关的基因组区域(图1)。
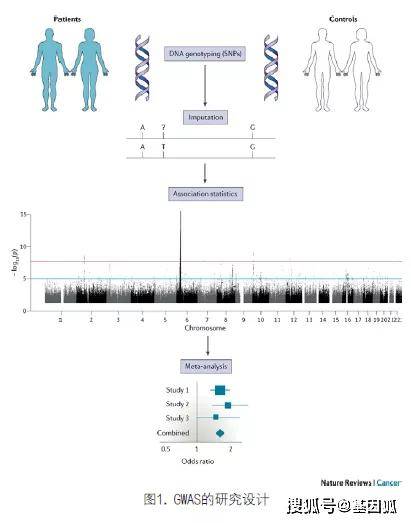
一般来说,GWAS需要适当的配对对照,从而确保足够的统计效能,并将导致假阳性关联的偏差或混淆因素最小化。为了抵消商业化SNP芯片的高成本并且保证统计效能,GWAS常基于分阶段策略而设计。随着芯片成本的降低以及国际联盟的形成,许多分析仅基于全基因组SNP数据的meta分析。通过使用“人类基因组单体型图计划”(HapMap计划)、“千人基因组计划”(1000 genome project)和UK10K联盟等项目提供的个人序列参考组合,对未定义的基因型进行推断,促进了不同列阵形式生成数据的统一。这使得频率低至0.1%的SNP等位基因得以精确推断,从而扩大了GWAS在破译癌症易感性等位基因结构方面的应用。
三、癌症风险位点的确定
在过去的十年中,欧洲人群包括乳腺癌、前列腺癌、肺癌、结直肠癌、胰腺癌、胃癌、肾癌和膀胱癌在内的每一种主要癌症都有许多GWAS报道。其中,还确定了许多东亚人和非洲裔美国人特定的风险位点,这反映了不同种族之间LD结构的差异。GWAS也用于恶性黑色素瘤、卵巢癌、基底细胞癌、神经胶质瘤、脑膜瘤、睾丸生殖细胞肿瘤、甲状腺癌和血液学的恶性肿瘤的研究,包括主要的B细胞肿瘤:急性淋巴细胞白血病,慢性淋巴细胞白血病、多发性骨髓瘤、霍奇金淋巴瘤、滤泡淋巴瘤和弥漫性大B细胞淋巴瘤。此外,通过GWAS发现了几种儿童实体癌的常见风险等位基因,包括Wilms肿瘤和神经母细胞瘤。目前,GWAS已在262个不同的基因组区域发现了超过430处的癌症关联。
乳腺癌和前列腺癌的GWAS发现了最多的风险位点。因为相关GWAS的样本量很大,每一个都涉及超过120,000人的基因分型,所以具有较大的统计效能。而对于其他癌症,遗传能力的差异可能影响了GWAS在识别风险位点方面的表现。例如,在具有强烈遗传性和8倍家族相对危险度(RR)的慢性淋巴细胞白血病中,GWAS仅基于6200名患者和17598名对照者发现了43个风险位点,相比之下,针对所有的肺癌亚型,GWAS在29266名患者和56450名对照者中仅发现了18个风险位点,这反映了非遗传风险因素在肺癌病因学中的重要性。
1.癌症风险位点的多效性:目前发现的大多数SNP关联都是癌症特异的,这与大多数家族性癌症风险的流行病学观察结果一致。然而,大约三分之一的SNPs位于与多种癌症相关的基因组位点。多效性位点是指某些特定热点与多种癌症相关,并且在不同类型中发挥作用的分子机制可能是相同的。多效性使癌症或位点分组成为可能,研究其共有机制或特征能够指导我们理解癌症。如,端粒相关的3q26.2位点(TERC)、5p15.33位点(TERT)、10q24.33位点(OBFC1,也称为STN1)和20q13.33位点(RTEL1)均与多种癌症风险相关。特别是,5p15.33位点的SNP rs2736100与神经胶质瘤、膀胱癌和肺癌风险相关。同样,9p21.3位点(CDKN2A-CDKN2B)被发现会影响胶质瘤、黑色素瘤、急性淋巴细胞白血病和肺癌的风险,还会影响获得性黑色素痣的密度,从而增加患恶性黑色素瘤的风险。对于某些位点,虽然涉及的SNPs不同,但最终的致癌机制是相同的。如8q24.21位点的SNP rs6983267是通过研究前列腺癌和结直肠癌发现的。此外,这个位点也被证明含有其他癌症的SNPs。这些SNPs定位在不同的LD区,通过调节MYC对癌症风险产生组织特异性影响(图2)。
多效性位点所涉及的癌症类型能够提供更多的生物学信息。例如,16q24.3与皮肤癌有多种关联,包括黑色素瘤、非黑色素瘤皮肤癌和皮肤鳞状细胞癌,这可能表明了一种常见的,可能是组织特异的作用机制。此外,多种癌症相关区域与已知的家族性癌症综合征相关,例如19p13.11区域与乳腺癌和卵巢癌相关。对于包含多个癌症关联的其他区域,共享的基因组位点可能是由于偶然性和完全独立关联的分子基础。探索多效性位点的性质可能是未来工作的重点,以对癌症易感性和病因学有更深入的了解。

绘制的为8号染色体部分区域:127,000,000—130,700,000;a:重叠的癌症关联信号;b:GWAS信号的相对位置;c:表观遗传标记——峰代表组蛋白修饰,表明DNA有可能影响邻近基因的基因表达,并通过环相互作用,影响远端基因的表达;d:该位点的RefSeq基因注释。
2.癌症生物学研究:GWAS有望通过在特定途径中识别基因变异,从而为癌症生物学提供新的见解。由于肿瘤生物学知识有限,阻碍了候选基因方法的发展,因此在先前的靶向关联研究中较少评估GWAS涉及的基因。此外,在GWAS的帮助下人们对不同癌症类型肿瘤发生的途径也有了新的认识;例如,B细胞发育和免疫应答基因(如IKZF1、CEBPE、IRF4、IRF8、GATA3和ARID5B)作为B细胞肿瘤风险的关键决定因素,其作用已经被确定。同样,GWAS也涉及参与发育转录调控、微管和染色体组装以及TGCT肿瘤发生过程中KIT-ERK信号通路组成部分的基因。
鉴于确定多种癌症致病因素存在相当大的困难,基因关联有可能支持当前的病因学假说,或提出需要通过基因特异性或环境特异性假说进行检验的新假说。当前有一些研究表明存在基因——生活方式交互作用,如位于15q25(CHRNA3–CHRNA5)位点的SNP,通过介导尼古丁上瘾间接地与肺癌风险相关。这个位点的基因型影响戒烟的能力,携带两拷贝CHRNA3-CHRNA5风险等位基因的吸烟者平均每天比那些非风险等位基因纯合子的吸烟者多吸两支烟。此外,位于8p22(NAT2)的SNP,它改变了吸烟对膀胱癌和皮肤癌相关的皮肤色素沉着点的影响。这些数据表明,遗传因素对癌症发病率的影响可能比之前认为的更大。
四、常见变异和遗传风险
通过GWAS识别的基因位点通常表现出剂量效应,风险等位基因纯合子携带者的风险大约是杂合子携带者的两倍。因此在GWAS研究中,常应用多基因模型进行分析。
目前发现的几乎所有的癌症易感性基因位点都与风险的适度增加有关,ORs通常小于1.5。也有一些例外,如骨髓增殖性肿瘤的9p21(JAK2)、TGCT的12q21.32(KITLG)和IDH突变胶质瘤的8q24.21(CCDC26)位点的SNPs,每一个位点都与相应肿瘤的三倍风险增加相关。值得注意的是,虽然这些癌症虽具有较大的家族风险,但几乎没有证据显示存在孟德尔遗传倾向。
这些GWAS数据为癌症易感性等位基因结构提供了共识。尽管与这些SNPs相关的癌症风险较低,但这些变异是常见的,因此它们都在很大比例的普通人群中增加了相应癌症的风险。常见变异(能解释超过1%的遗传风险)的数量非常少。然而,由于GWAS识别的SNPs必须通过一个非常严格的显著性阈值,因此可能存在一些低于阈值的常见变异,虽然不符合显著性标准,但仍然导致了特定癌症的遗传风险。所以,量化已知和潜在的易感性SNPs解释的遗传能力,对于验证癌症的病因学基础和了解其遗传结构具有重要意义。计算由大量复杂人类疾病相关的SNPs解释的表型变异比例是一个相当大的挑战。全基因组复杂性状分析(GCTA)显示常见变异可能解释了许多癌症的大部分遗传风险,如雌激素受体阴性乳腺癌为10%,前列腺癌为38%,结直肠癌为17%。近年来,人们尝试通过分析小等位基因频率、LD和基因型的不确定性来完善GCTA,从而提高了对常见变异遗传力的估计值,因此目前多基因遗传对癌症遗传风险的贡献可能被低估。
五、破译风险位点
GWAS揭示了LD中与tagSNP高度相关的功能变异的影响。因此很难确定在一组紧密连锁的基因位点中,哪一个是与疾病相关的功能性变异。虽然少数GWAS的tag SNP是直接起作用的,如与结直肠癌相关的8q24.21(rs6983267)。但是大多数tag SNP可能只是与致病SNP存在LD关系。通过推断实现精细定位,能够在帮助进一步确认致病SNP。此外,精细定位也可以解析关联信号,例如通过对胶质瘤易感区域8q24.21的精细定位,发现SNP rs55705857足以解释此前认为是相互独立的两个tag SNP信号。
功能性的遗传变异是GWAS识别的风险位点的基础。由于一些等位基因型变化会影响基因表达的分子机制,因此从这个角度考虑将有助于筛选GWAS数据。到目前为止,只对小部分风险位点进行了全面研究,关于常见变异介导癌症易感性的遗传生物学基础研究仍在进行中。GWAS鉴定出的少数位点直接影响表达蛋白的氨基酸序列,继而影响蛋白功能。比如与肺癌和乳腺癌易感有关的BRCA2p.Lys3326Ter(rs11571833)和CHEK2p.Ile157Thr(rs17879961);位于TP53 3ʹ非翻译区(ploy A尾部)的SNP(rs78378222),通过影响RNA的加工,介导前列腺癌和胶质瘤风险;以及5p15.33处的抑制性剪接异构体rs10069690变异,会影响剪接位点,从而导致端粒酶活性降低。然而,编码变异除扰乱蛋白质功能外,也可能作为功能性的非编码变异的标签。
由于非编码区约占基因组的99%,所以GWAS鉴定的大多数风险位点位于基因组的非编码区(例如,基因内含子或启动子和基因间区),而且这些低外显型的风险多态性具有比直接影响氨基酸序列更精细的调控作用。
GWAS鉴定的风险位点定位于细胞类型特异性活性染色质的基因组区域,并且多是定量性状位点,甲基化定量性状位点和转录因子(TF)结合相关位点。染色质构象研究有助于将SNPs定位的调控区与其各自的靶基因联系起来。有研究证明,拓扑相关结构域内增强子和启动子区域之间的染色质环相互作用所介导的顺式调控效应可能是许多GWAS信号的功能基础。
为了解癌症风险位点的调节机制,人们已经做出了巨大的努力。其中基于汇总数据的孟德尔随机化和其他统计学方法,以及提供公开的调控区域图谱的ENCODE,NIH Roadmap Epigenomics和BLUEPRINT epigenome等计划都发挥了相当大的作用。此外,基于网络的方法能够了解控制疾病易感性的高层次结构。例如,特定TF的结合可以在风险位点处富集,而此类TF在肿瘤中经常发生突变并具有相关的生物学活性。
8q24.21区域是GWAS中出现的最有趣和最重要的位点之一,也是这种调控机制的一个很好的例子。在同一拓扑相关区域128至130Mb的基因组间隔内包含具有大肠癌、胶质瘤、慢性淋巴细胞白血病、多发性骨髓瘤、霍奇金淋巴瘤、前列腺癌、乳腺癌和膀胱癌特异性的多个独立位点(图3)。其中与大肠癌和前列腺癌相关的8q24.21SNP rs6983267位于进化保守区域,rs6983267的两个等位基因变体和TCF7L2差异结合,并且该区域作为转录增强子可以与原癌基因MYC相互作用。最近对该区域的Hi-C分析显示了一种更复杂的调控机制,发现各种大的基因间非编码RNA介导了风险位点的效应(例如,结肠癌相关转录本1(CCAT1)、前列腺癌相关转录本1(PCAT1)和CCDC26分别与大肠癌、前列腺癌和胶质瘤发病风险相关)。目前8q24.21与各种癌症风险的调控机制研究还处于相对初级阶段,今后的深入研究可能会利用模型系统探索组织特异性效应,以及利用CRISPR-Cas9干扰候选调控元件。
SNP与亚型特异性。随着研究深入,在许多癌症亚型中,SNP基因型与癌症表型的关系越来越显著。比如位于5p15.33(TERT-CLPTM1L)和3q28(TP63)的SNPs显著影响肺癌组织学,主要与腺癌相关,而13q12(BRCA2)和人类白细胞抗原的关联是肺鳞癌特有的。类似地,许多胶质瘤风险位点具有亚型特异性,例如5p15.33、20q13.33和7p11.2与胶质母细胞瘤发生有关,11q23.3和8q24.21与非胶质母细胞瘤的发生有关。
迄今为止,表现出最显著基因型-疾病表型关系的SNP是与急性淋巴细胞白血病发病风险有关的10q21.2(ARID5B),它选择性地作用于ALL患者的超二倍体性B淋巴细胞前体亚群。此外,cyclin D1(CCND1)c.870G>A SNP特异地与具有(11;14)(q13;q32)易位的骨髓瘤相关。研究推测,这种亚型特异性的关联反映了特定的突变信号环境,从而有助于进一步深入了解肿瘤的发展。相对于非风险等位基因,与癌症风险增加有关的易感等位基因可能具有选择性优势,从而在特定的癌症中优先富集。近期研究发现,位于10q21.2的SNP rs709044在具有超二倍体性的原始细胞中更容易保留,这与导致正常淋巴细胞发育停滞的遗传变异一致,继而促进白血病性的克隆扩增。同样,错义变体CDKN2A p.Ala148Thr(rs3731249)的风险等位基因也具有选择优势,与急性淋巴细胞白血病发病风险增加有关。
最近的复杂疾病易感性的全基因模型(omnigenic model)提出,疾病相关组织中的任何调控变异都会对疾病风险产生影响。在这个模型中,如果基因在疾病病因学中具有特定作用,则将其定义为“核心基因”,如果其作用是间接的,则将其定义为“周围基因”。考虑到周围基因比核心基因更多,以及所观察到的影响范围,研究认为疾病的总遗传贡献中有很大一部分来自于在疾病中不起直接作用的周围基因。因此,在这个模型中,周围基因通过网络以相对精细的方式影响核心基因的调节和功能。但此模型是基于当前对癌症和网络生物学的有限理解,因此有待实验进一步证明。

a:A>G多态性改变TF结合,通过环状启动子-增强子-复合体相互作用影响基因转录;b:A>G多态性发生在内含子剪接位点并导致内含子保留,从而影响mRNA的加工;c:A>G多态性导致在LincRNA上产生一个新的microRNA结合位点;d:A>G多态性通过引起酪氨酸变成胞嘧啶的氨基酸替换来影响蛋白质序列。
六、GWAS的临床相关性
癌症遗传学不仅可以应用于风险分层,而且有助于更好地理解癌症发展的基础。在许多情况下,这些信息可以直接应用于临床(图4)。
1.药物发现与重新定位:癌症基因组测序研究表明GWAS涉及的调控区和靶基因经常发生体细胞突变,这反映了它们的驱动活性。这类研究有助于破译风险位点,助力药物研发。实际上,GWAS已为许多成功获批的药物提供了直接的支持性遗传证据,助力药物开发和潜在的药物重新定位。例如,GWAS确定IL-23信号通路为银屑病发生的危险因素,并且IL23R编码基因的p.Arg381Gln(rs11209026)多态性能够保护人体免受多种炎症性疾病的侵袭。而ustekinumab作为一种单克隆抗体,可以中和IL12和IL23共享的p40亚单位,为银屑病患者的治疗提供了新的选择。此外,抑制慢性淋巴细胞白血病的BCL2和抑制乳腺癌的成纤维细胞生长因子受体(FGFR)也是与GWAS相关的癌症治疗方法。当前仍需要进一步的工作来识别GWAS相关的靶基因和异常生物学途径,并确定胚系和体细胞突变,以最大限度地发挥GWAS在药物发现中的潜力。
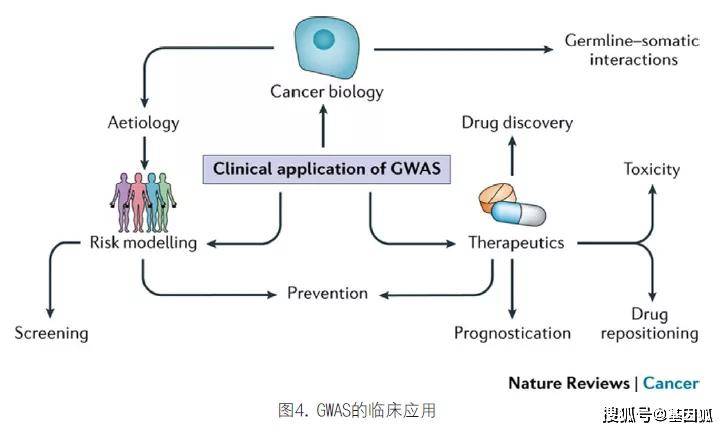
如本文所述,GWAS除了拓展对癌症生物学的了解外,还可以提供癌症病因学风险因素的信息。通过风险建模,来自GWAS的数据可以帮助识别患癌症风险增加的个人,从而帮助预防癌症,并通过筛查改善早期发现。通过GWAS确定的基因可以为药物发现和重新定位提供信息,并为癌症预后和治疗相关并发症的提供指导。
2.分层筛选:根据基因型识别高危人群有助于定制预防或筛查策略,具有深远的临床意义。由GWAS发现的大多数癌症风险变异仅轻度增加患病风险,这使得人们一直认为SNPs难以走入临床。然而,单个SNPs相关的小效应仍有临床应用潜力。一项关于结直肠癌以及乳腺癌和前列腺癌的研究证明,多个风险SNPs的综合效应有可能在一定程度上辨识风险,有助于疾病的预防和筛查。在该研究中,基于37个已知的结直肠癌风险变异的多基因风险评分(PRS)表明,与人口中值相比,得分前10%的个体患结直肠癌的风险将增加1.8倍,而排在前1%的个体患结直肠癌的风险将增加2.9倍(图5)。该研究提示利用PRS有可能提高筛查效率,以便及早发现结直肠癌、前列腺癌和乳腺癌。此外,PRSs的风险鉴别可能为制定和实施化学预防策略提供信息。近期一项预测BRCA1和BRCA2突变携带者的乳腺癌和卵巢癌风险的研究提示:PRSs在评估癌症易感基因携带者的患癌风险方面具有应用潜力。
3.指导预防:孟德尔随机化法(MR)利用GWAS数据可识别非遗传危险因素和潜在化学预防剂。例如,通过使用遗传标记作为高脂血症的工具变量(即遗传工具),研究高胆固醇血症和结直肠癌之间的因果关系。SNPs降低3-羟基3-甲基戊二酰辅酶A还原酶(HMGCR)表达,这与他汀类药物降低胆固醇的机制相似,而这些SNPs的遗传风险评分与降低结直肠癌风险相关,因此提示他汀类药物不仅对冠心病治疗有作用,对于结直肠癌人群也有益处。但是进行MR分析的一大挑战是:遗传变异作为有效工具变量需要满足无多效性假设。虽然在这类研究中已经开发出量化多效性的方法,但传统的基于MR的分析中不易检测到网络多向性。(即单个变体可能会影响多个性状,但这些性状并不是因果相关的,而是通过相同细胞类型中的相同调控网络介导的)。
4.指导治疗:胚系突变作为一种潜在的预后因素奠定了肿瘤发生发展过程中个体间差异的基础。研究发现,药物代谢相关基因变异与慢性淋巴细胞白血病、肺癌和乳腺癌的预后有关。因此,对于CYP3A7*1C携带者来说,以细胞色素P450 3A(CYP3A)为底物的环磷酰胺、紫杉烷和米托蒽醌不是最优的化疗方案。此外,GWAS可成功识别有治疗相关毒性风险的个体,如蒽环类药物引起的心脏毒性和放射导致的组织损伤。GWAS通过识别影响药物疗效和药物毒性的常见基因变异,从而实现个性化用药,改善治疗决策。
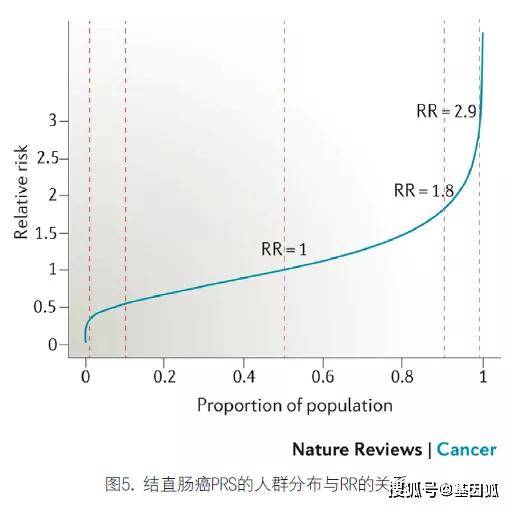
垂直红线(从左到右)分别对应于百分位数1、10、50、90和99。与人群中位数相比,得分前10%的个体患结直肠癌的风险将增加1.8倍,而排在前1%的个体患结直肠癌的风险将增加2.9倍。
七、总结和展望
GWAS已经证明,多数常见癌症的大部分可遗传风险是多基因的。因此,癌症遗传易感性是由人群中不同风险和患病率水平的易感等位基因组合而成的。除了乳腺癌和前列腺癌之外,目前确定的位点仅解释了许多癌症的小部分家族性风险。许多GWAS结果包括低OR关联的位点,这表明更大规模的研究应该会识别更多新的易感基因位点。虽然罕见的复发性致病变异可能不会对癌症的可遗传风险做出实质性贡献,但对这类风险变异的发现仍不足。使用最近开发的参考组合对GWAS数据进行推断,从而识别亚多态风险等位基因(即风险等位基因频率<1%),这可能有助于发现罕见的复发性致病变异。
GWAS确定的位点极大地扩展了现有的影响癌症风险的基因谱系,然而确定GWAS数据中的功能序列仍具有挑战性。为了更好地了解癌症生物学,并为治疗和预防策略提出潜在的目标,必须充分利用GWAS。利用如CRISPR–Cas9等模型系统和策略的发展将会有助于确定功能序列。
本文译自《Nature Reviews Cancer》
编译:杨子瑶 刘芃菲
校审:赵晓涛
单位:北京大学人民医院检验科
本文来源于《临床实验室》2020年第5期“肿瘤及伴随诊断”专题
- 本文固定链接: https://maimengkong.com/zixun/1502.html
- 转载请注明: : 萌小白 2023年5月1日 于 卖萌控的博客 发表
- 百度已收录
