使用R语言clusterProfiler包进行KEGG富集分析(有参分析)
我们在前面的视频教程中,为大家讲述了GO、KEGG富集分析的基本原理,并展示了如何使用DAVID进行GO富集在线分析的过程,详情请添加微信公众号查看“没想到基因GO富集分析这么简单”。本节继续以R语言clusterProfiler包中的方法为例,展示如何进行KEGG富集分析。下文使用的测试数据和R代码等,可添加微信公众号获取。
以下是图文版的简化教程:
1 准备输入数据
我们提供的示例数据为人类基因,文件中共包含两列。第一列是基因的ENTREZ ID,用于和KEGG数据库建立联系对应基因以便进行富集分析;第二列是基因的SYMBOL ID,也就是通俗的基因名称,用于基因名称转换用,详见下文过程或上文视频。
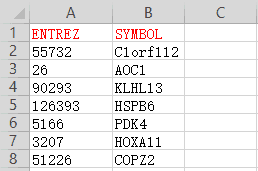
注:clusterProfiler默认只能识别3种类型的基因名称用作富集分析,ENTREZ ID、NCBI ID以及UniProt ID。我们的示例文件中就使用的ENTREZ ID。如果老师或同学们不清楚怎样转换基因ID,例如ENSEMBL ID向ENTREZ ID的转换,可以添加微信公众号,联系我们寻求帮助。
2 clusterProfiler的KEGG富集分析
读取基因列表文件至R中,加载并使用clusterProfiler的内部函数enrichKEGG()即可自动完成KEGG富集分析。
注:以下方法均对于KEGG数据库中已经收录的物种而言的,若目标物种未包含在KEGG数据库中,则无法进行KEGG富集。例如该示例的物种是人类,其它物种查询可点击以下链接:
https://www.kegg.jp/kegg-bin/get_htext?htext=br08601_ko00001&query=ko&option=-w
!!!**************************************************
#bioconductor 安装 clusterProfiler
#install.packages('BiocManager') #需要首先安装 BiocManager,如果尚未安装请先执行该步
#BiocManager::install('clusterProfiler')
#加载R包
library(clusterProfiler)
#读取待富集的基因名称(示例数据,基因名称列表)
gene_list <- read.delim('gene_list.txt', stringsAsFactors = FALSE)
#每次打开R计算时,他会自动连接kegg官网获得最近的物种注释信息,因此数据库一定都是最新的
kegg <- enrichKEGG(
gene = gene_list$ENTREZ, #指定基因名称,例如 ENTREZ ID
keyType = 'kegg', #指定基因名称类型,kegg 代表了使用 ENTREZ ID
organism = 'hsa', #hsa代表人类,其它物种更改这行即可,可在 KEGG 官网查询物种名缩写
pvalueCutoff = 1, #p 值 p 调整值之类的,指定 1 为阈值,也就是将所有的条目都输出了
qvalueCutoff = 1,
pAdjustMethod = 'fdr' #p 值调整方法
#输出,默认情况下基因名称显示为富集时使用的名称
write.table(kegg, 'KEGG.ENTREZ.txt', sep = '\t', quote = FALSE, row.names = FALSE)
!!!**************************************************
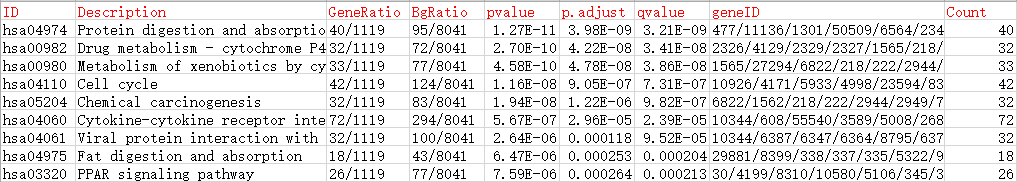
最后输出富集分析结果,对于各列内容:
ID和Description,富集到的KEGG条目的名称和描述;
GeneRatio和BgRatio,分别为富集到该KEGG条目中的基因数目/给定基因的总数目,以及该条目中背景基因总数目/该物种所有已知的KEGG功能基因数目;
pvalue、p.adjust和qvalue,富集p值、校正后p值和q值信息;
geneID和Count,分别为富集到该KEGG条目中的基因名称和数目,由于分析中使用的ENTREZ ID,故这里也默认显示为ENTREZ ID。
3 基因名称转换
由于ENTREZ ID只是一串数字编号,不利于辨认,因此我们可以考虑将它转换为通俗的基因名称,也就是使用SYMBOL ID。
!!!**************************************************
#基因名称转换,例如将基因 ENTREZ ID 转换为 SYMBOL ID
kegg <- read.delim('KEGG.ENTREZ.txt', stringsAsFactors = FALSE)
rownames(gene_list) <- as.character(gene_list$ENTREZ)
for (i in 1:nrow(kegg)) {
new <- c()
old <- unlist(strsplit(kegg[i,'geneID'], '/'))
for (id in old) new <- c(new, gene_list[id,'SYMBOL'])
kegg[i,'geneID'] <- paste(new, collapse = '/')
}
write.table(kegg, 'KEGG.SYMBOL.txt', sep = '\t', quote = FALSE, row.names = FALSE)
!!!**************************************************
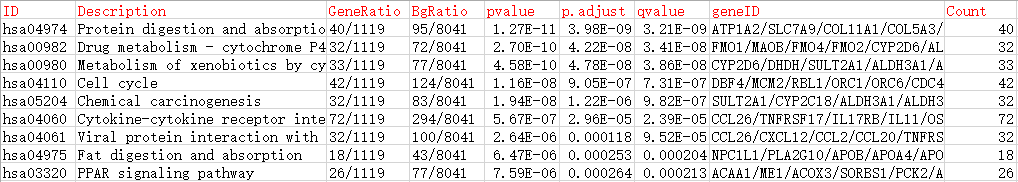
新输出的表格中,geneID中的基因名称就转换为SYMBOL ID了,这样我们就可以很清楚地知道富集条目中所包含的基因具体是哪种类型的。
4 clusterProfiler附带的作图功能
此外,clusterProfiler中也额外提供了一系列的可视化方案用于展示本次富集分析结果,具有极大的便利。
!!!**************************************************
#clusterProfiler 包里的一些默认作图方法,例如
barplot(kegg) #富集柱形图
dotplot(kegg) #富集气泡图
cnetplot(kegg) #网络图展示富集功能和基因的包含关系
emapplot(kegg) #网络图展示各富集功能之间共有基因关系
heatplot(kegg) #热图展示富集功能和基因的包含关系
!!!**************************************************
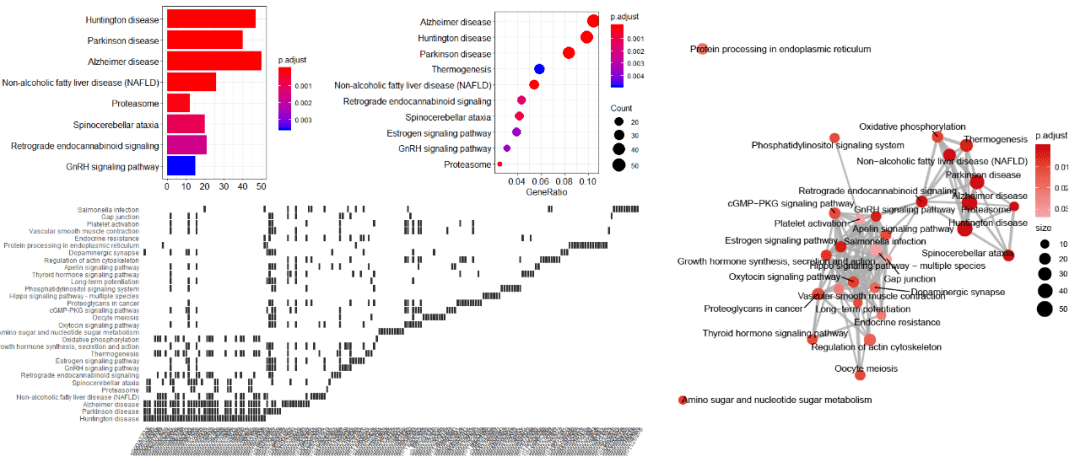
这样,KEGG富集分析结果表连同富集柱形图、气泡图等一起就都获得了,是不是很快捷方便呢?
此外,若老师或同学们有RNAseq(mRNA、lncRNA、miRNA、circRNA)或蛋白质组等数据分析、绘图等问题疑问,欢迎大家来电咨询。
!
!
!
注:!!!*******之间为R脚本内容。
搜索微信公众号“纪伟讲测序”- 本文固定链接: https://maimengkong.com/image/1016.html
- 转载请注明: : 萌小白 2022年6月23日 于 卖萌控的博客 发表
- 百度已收录
