目前,转录组测序仍是应用最广的高通量测序技术之一,很多研究课题是关于基因表达潜在的机制,并已经发现了一些现象,但分子机制还不清楚。而做转录组测序特别适合用于分子机制探究,可以获得样本中几乎所有的mRNA信息。关于转录组领域的研究,应用范围极为广泛。如可研究同一个体不同组织之间的基因表达差异;或者不同的外界处理条件下(病毒、光照、紫外、干旱、高温和高盐胁迫等),对基因表达的影响。
在我们正式进行转录组数据分析之前,需要先对组内生物学重复(一般设置3个生物学重复)进行样本关系分析,判断组内重复性效果的好坏,是否有离群样本。应广大研究者之需,本期针对大家比较关心的样本重复性问题进行探讨,力争为各位老师在科研之路上带来帮助。
在进行问题讨论之前,首先我们对可能会困扰大家的关于什么是生物学重复和技术学重复的问题进行区分。
①生物学重复:指同一处理下不同的生物学样品。由于遗传和环境等因素的影响会引起生物体的个体差异,因此需要采用生物重复的实验设计方法来降低该差异。一般的实验设计中,都会包括实验组和对照组。如下图A实验组包含3只小鼠,那么这3只小鼠,经过相同的实验处理,分别测组织的RNA-seq,即为一组生物学重复。
②技术重复:简单来说就是对同一生物体样品进行重复地检测。如下图B、C,都属于技术重复。对于第一种技术重复,重点是检测RNA-seq方法的准确度。比如当发现了一个新的检测基因表达量的方法,就需要用这种重复来验证(图1 B);第二种技术重复重点是这个小鼠本身的基因表达水平(图1 C)。
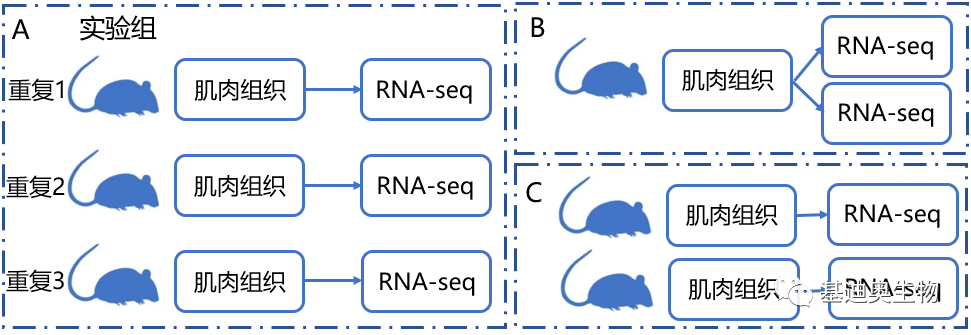
图1 生物学重复和技术重复
那么接下来,我们正式切入主题,针对样本重复性问题进行探讨。
『1. 生物学重复必须要设置吗?』
答:需要。生物学实验中,生物体往往存在异质性,常常需要设置重复,以此确保不是个体的偶然变异对结果产生的影响[1]。若不设置组内生物学重复,在投稿时也会受到审稿人的质疑。我们无法判断组内差异所占的比例有多大,可能获得的差异表达基因仅仅是少数个体差异的表现,并不能反映是真正处理效应导致的差异。设置生物学重复可以评估组内误差,降低背景差异,检测离群样本,增强结果的可靠性。
Tips
组间差异是由组内差异和处理效应共同导致的[2] 。组内差异包括采样个体间的差异、实验操作误差等等,这些差异是我们在实验时要尽可能降低的。而组内误差主要由生物学误差和技术误差引起的。
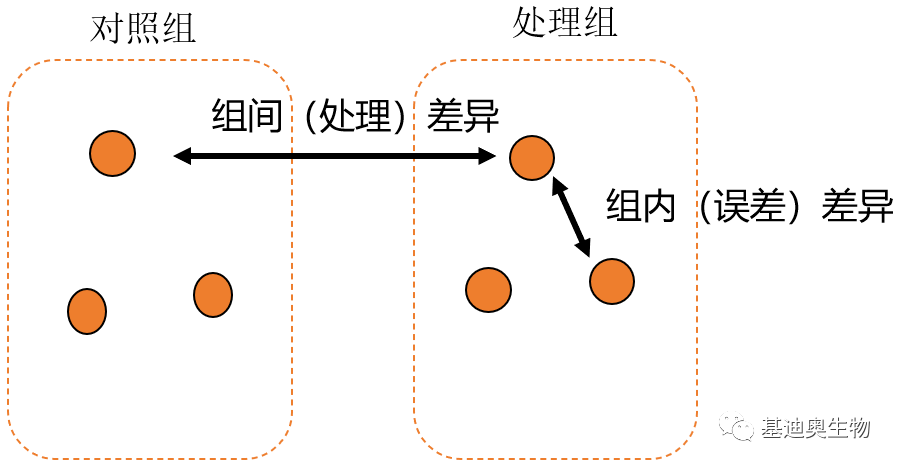
图2 组间差异和组内差异
『2. 每个处理推荐多少个生物学重复呢?』
答:不同的实验样品,由于外界因素导致的个体之间的差异或实验操作导致的误差可能不同。因此,针对不同的样品所推荐的组内生物学重复也有所差别 [3] 。
① 对于动植物样品,建议3~5个生物学重复,对生物学样品之间做相关性检验,提高实验结果的可信度;
② 对于细胞样品,生物学重复之间的差异性相对较小,建议3个以上生物学重复;
③ 对于临床样品,由于供试者的基因型、生活方式、生活环境、年龄、性别可能存在较大差异,可能需要更多的生物学重复,一般10个生物学重复以上 [4] 。
Tips
在转录组测序时,一般不建议设置两个重复。因为如果两个重复样品结果不一致,无法确定以哪个数据为参考。
『3. 用于判断组内重复性好坏的常用工具有哪些?』
答:在实际分析过程中确认组内重复性的好坏方法有很多,可进行样本的PCA,可计算两两样本的相关系数,或者绘制样本聚类图、重复性散点图多种方式综合判断。在实际分析中,通常结合PCA和相关性系数综合判断样本是否离群。
① PCA:详见Question 4;
② 相关系数:通常计算两个样品之间的Pearson或Spearman相关系数判断组内重复性情况。相关系数越接近1,样品间相似度越高。一般情况下,组内生物学样本相关系数大于组间样本,则表明组内重复性较好;
③ 样本聚类树:可用于判断在不同实验条件下的表达模式。依据样品的表达谱进行聚类,样品之间重复性较好时通常会聚在同一分支下。如果组内样本重复性较差可能会呈现无规则的聚类形式;
④ 重复性散点图:展示组内样本的重复性情况。图中偏离对角线的点越少,样品间的相关性越高,重复性越好。
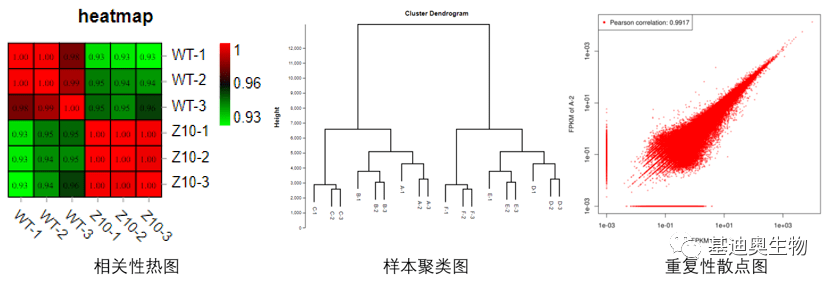
图3 Omicsmart中样本关系分析图形
『4. PCA是什么?怎么看?』
答:主成分分析(Principal Component Analysis,PCA)是一种线性降维算法。用方差(Variance)来衡量数据的差异性,将高维数据用某几个综合指标来表示。将原本鉴定到的所有基因的表达量重新线性组合,形成一组新的综合变量,同时根据所分析的问题从中选取2-3个综合变量,使它们尽可能多地反映原有变量的信息,从而达到降维的目的。如PC1(Principal Component 1)和PC2(Principal Component 2)为降维后获得的两个主成分因子,可分别从数据差异性最大和次大的方向提取出来。
在样本关系分析过程中,PCA可以让我们非常直观地看出各个样本之间的相似性。关于转录组测序,我们可能获得上万个基因的表达信息,那么利用PCA可将样本所包含的上万个维度的信息(上万个基因的表达量),降维至某些维度的综合指标(主成分)表示。一般选取PC1和PC2,来解释样本间的重复性好坏与组间样本的差异度。如下图PCA散点图,组内样本呈现相互聚集,说明组内的重复性比较好。
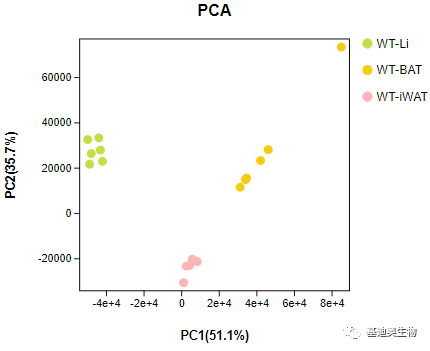
图4 Omicsmart在线报告PCA图
Tips
在文章中,也会看到三维的PCA图。这时选取了PC1,PC2,PC3去解释样本间的距离。PC1+PC2(+PC3)越大,对方差解释度越大,越具有说服力。
『5. 相关性系数分析时,相关系数达到多少可认为组内重复性效果好?』
答:一般情况下,计算相关性系数时,对于生物学重复(如采样时个体差异)之间的相关系数依据经验建议在0.7以上较好;对于技术重复(实验操作、实验仪器等)之间的相关系数依据项目经验来说在0.85以上比较合理。
Tips
关于相关系数如何计算,可能还存在不少的困惑。我们在这里也解释一下。对于转录组数据,可以利用样本的表达谱来计算样本间的相关性,通过计算相关系数r来评估每组样本的生物重复性。最常用的度量是Pearson和Spearman相关系数。
那么在实际分析中,这两种计算方式应该如何选择呢?
我们首先简单了解二者的区别。对于Pearson相关系数很简单,主要用来衡量两个数据集的线性相关程度。而Spearman相关系数它不关心两个数据集是否线性相关,所关注的是单调相关。所以Spearman相关系数也称为等级相关或者秩相关(即rank)。从下图中我们可以更好的理解,如果对数据进行线性变换(y=ax+b;a≠0),两者相关系数的绝对值都不会发生变化(图5 A);如果对数据进行单调但不是线性的变换,比如最常见的log scale,Spearman相关系数的绝对值也不会发生变化 [5] (图5 B)。 这时我们就可以知道,两者的前提假设就不同,Pearson相关假设数据集在同一条直线上,而Spearman只要求单调递增或者递减,所以Pearson的统计效力一般情况下比Spearman要高。但是更重要的是,我们需要根据实际情况选择正确的假设。比如,某个实验做了3次生物学重复,那有理由假设这3次重复线性相关。而如果是一个基因和另一个受到调控的基因的表达水平,或者某个基因顺式作用元件的染色质开放程度,和这个基因表达水平之间的关系就可能需要假设单调相关。
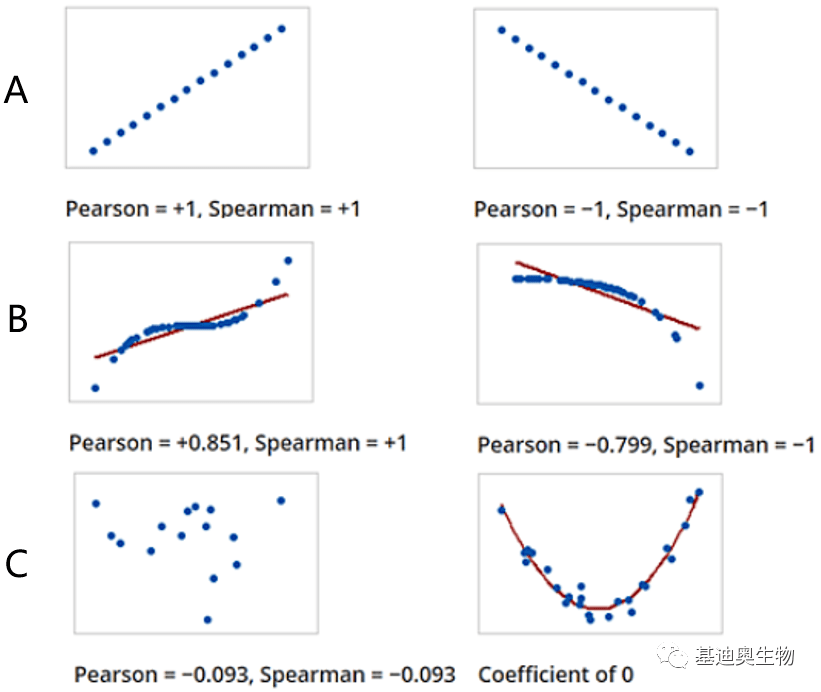
图5 Pearson和Spearman相关系数
关于两者的特点也有所不同,若想要深入学习二者的算法特征,可回顾往期文章 《相关系数第一弹:哪哪都能看到的皮尔森相关》 和 《相关系数第二弹:斯皮尔曼相关》 ,都有详细的解释哟。
『6. PCA和相关系数的算法,哪个更能判断样本的重复性?为什么?』
答:相关系数。因为PCA为把对样品贡献大的信息保留,所描述的是整体所有组的特征;而相关系数直接呈现的是两组样品之间的相关程度。若相关系数越高,表明两组样品之间的相关程度越高,即重复性越好。
『7. 样本离群了,还能用于分析吗?』
答:首先判断离群程度,若离群程度较小,则可以尝试设置阈值,缩小基因范围,再次重新进行相关性分析判断样本是否离群。若离群程度很大,对后续差异分析的结果造成了很大的影响,那么可以考虑将该样本剔除,再进行后续差异分析等等。
Tips
转录组测序通常要求设置3个生物学重复样本,如果样本足够多,建议比预期实验设计多送1~2个样本测序,以便后续某个样品与组内其它样本出现离群情况,直接剔除离群样本,省时省力。若测序样本较少,无法剔除样本,也可以考虑对同一批次的备份样本再次测序,后续再重新分析。
以上就是今天的关于样本关系分析问题,在此也向广大研究者征集相关问题,如有疑问,欢迎下方留言。或者也可登录基迪奥OmicShare论坛,搜索和讨论更多相关知识。
论坛网址:
https://www.omicshare.com/forum/
▼
参考文献
▼
[1] Robles, José A et al. Efficient experimental design and analysis strategies for the detection of differential expression using RNA-Sequencing. BMC genomics vol , 13 484. 17 Sep. 20 12, doi:10.1186/1471-2164-13-484
[2] Hansen, K., Wu, Z., Irizarry, R. et al. Sequencing technology does not eliminate biological variability. Nat Biotechnol . 29, 572–573. 2011, https://doi.org/10.1038/nbt.1910
[3] Todd E V, Black M A, Gemmell N J. The power and promise of RNA-seq in ecology and evolution[J]. Molecular ecology, 2016, 25(6): 1224-124 1
[4] Liu Y, Zhou J, White K P. RNA-seq differential expression studies: more sequence or more replication?[J]. Bioinformatics , 2013, 30(3): 301-304
[5] Trost B, Moir CA, Gillespie ZE, et al. Concordance between RNA-sequencing data and DNA microarray data in tranome analysis of proliferative and quiescent fibroblasts. R Soc Open Sci . 2015, 2(9):150402. doi:10.1098/rsos.150402
- 本文固定链接: https://maimengkong.com/zu/992.html
- 转载请注明: : 萌小白 2022年6月17日 于 卖萌控的博客 发表
- 百度已收录
