上期我们分享了关于样本重复性问题的一些小技巧 ,在我们突破第一关,做好样本关系分析后,这时在转录组测序获得的茫茫基因中,该如何找到心仪的目标基因呢?毕竟我们真正关心的是与处理效应相关的基因,而差异表达的基因才往往是造成组间差别的驱动因素。因此我们需要特定的方法找出这些差异基因,这种方法就是差异分析。
本文针对在做差异分析时大家可能经常存在的疑点,如为什么是基于负二项分布?以及进行差异分析时软件该如何选择等问题进行整理,力争为各位老师在科研之路上带来帮助。
术语介绍
首先我们简单介绍二代测序中经常听到的reads和read counts:
Reads:指测序得到的每一个片段称为一个reads。
Read counts:比对到一个基因上的reads数目。
二代测序因读长的限制,要先把mRNA随机打断,以片段化的mRNA为模板反转录cDNA后进行测序,读取的一段段序列称为reads。如PE150,PE指的为paired-end,双端测序,150指两端各测到的碱基数。
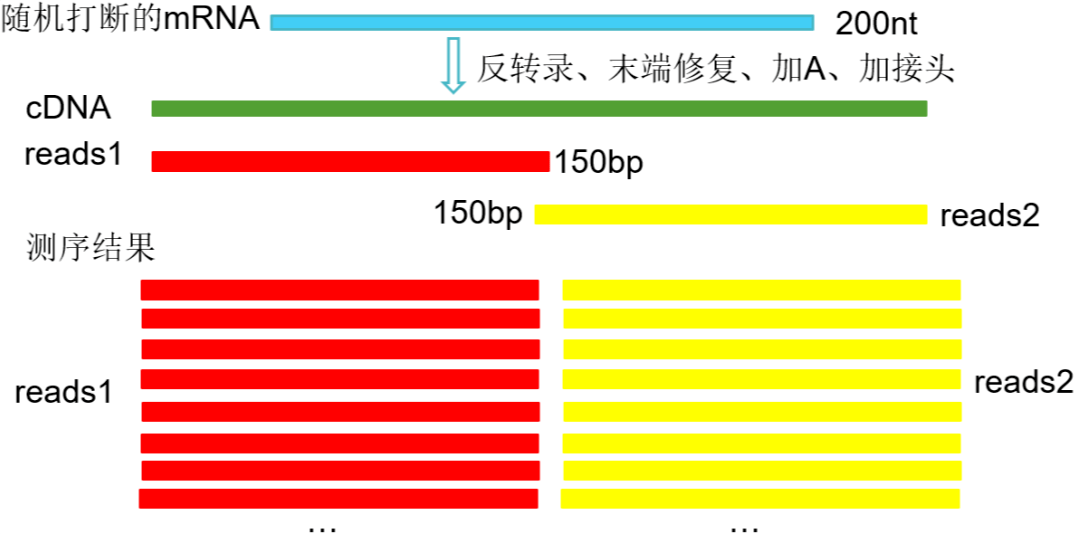
图1 Illumina测序(PE150)
*注:这里是将mRNA先打断,再反转录成cDNA。若先反转录mRNA,再进行cDNA的片段化,对后期转录本有不同的偏好性。先针对mRNA进行打断再反转录获得测序reads主要是针对基因本体;若先反转录,再打断测reads将对转录本3’端具有较强的偏好性(图2),因此目前测序一般为先对mRNA打断再反转。
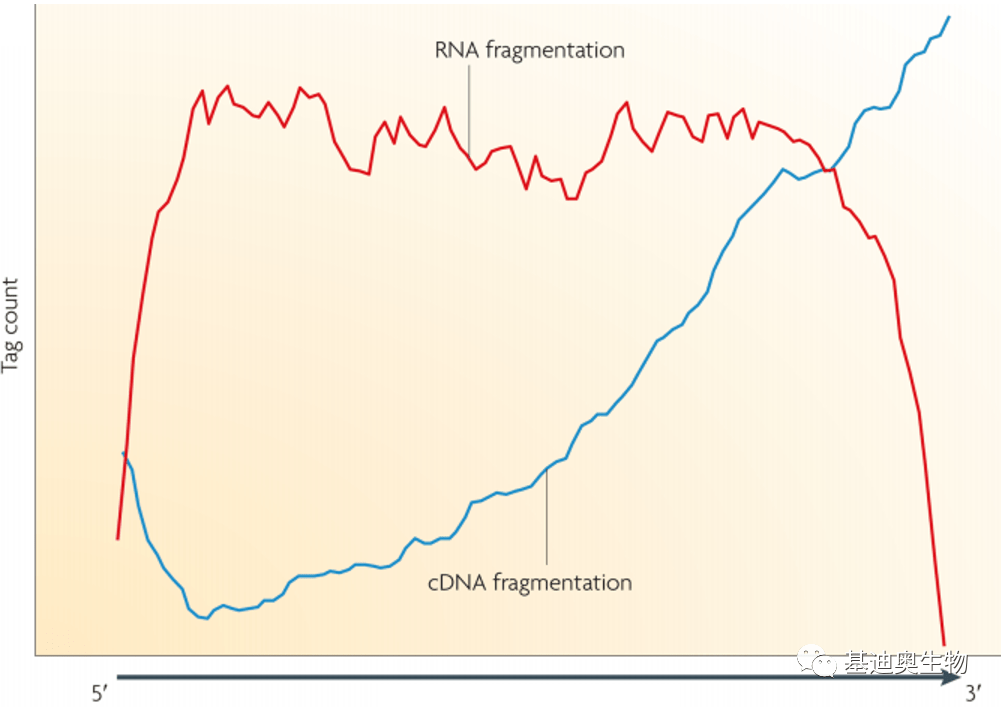
图2 不同方法建库对后期读数的偏好性
红线:RNA片段化;蓝线:cDNA后片段化
差异分析相关问题解析
Q1:为什么做差异表达的时候是基于负二项分布而不是正态分布,和泊松分布有什么区别?
答:正态分布是连续数据的一种理想状态,但是对于转录组测序得到的数据,通常实验样本数较少(n<20),且任何基因的表达量不为负数,用于表征表达量的counts数也是非连续的,这些数据并不符合正态分布。那么为什么现在做差异分析普遍基于负二项分布呢?在评估为什么采用负二项分布之前,首先简单了解涉及统计学的一些相关概念:
①二项分布:从n个独立试验中,得到成功事件k的分布;
②泊松分布:可由二项分布转化而来。在n个独立试验(可以是无限)中,k个事件发生的概率很小时常使用泊松分布;
③负二项分布:指所有到r次成功时即终止的独立试验中,失败次数为k的分布。其为一种离散概率分布,可以在泊松分布的基础上指定dispersion(离散程度)。
那么,用于差异分析的原始read counts数据适合哪种分布呢?
对于RNA-seq数据,通常往往获得大量的RNA序列,而提取特定转录本的可能性非常小,这种情况与泊松分布适用情景比较吻合。在转录组数据分析的早期,确实有学者采用泊松分布进行差异分析,但是发展到现在,几乎全部基于负二项分布,原因主要考虑到我们数据中均值和方差的关系。
对于泊松分布而言数据特点“均值=方差”,但真实的counts数据并不符合这样的规律,方差往往会大于均值(图3)。因此需要指定dispersion来描述方差偏离均值的程度,而负二项分布可在泊松分布的基础上指定dispersion,可作为描述总体的分布。
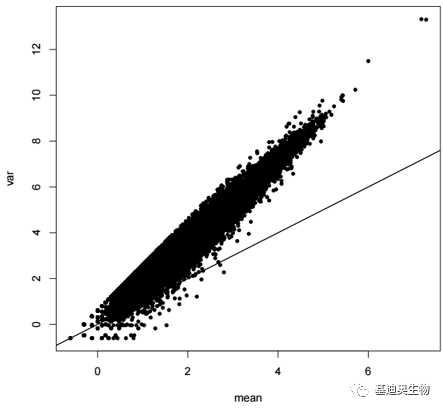
图3 估计的均数与方差[1]
Q2:差异分析时所用的edgeR和DESeq2是什么?该如何选择?
答:这两个均为目前做差异分析时比较主流的软件,是基于负二项分布[2-3]进行分析的R包。为了方便大家更好理解这两种软件,以表格对比的形式总结了两种软件的一些使用信息。
表1 edgeR和DESeq2比较
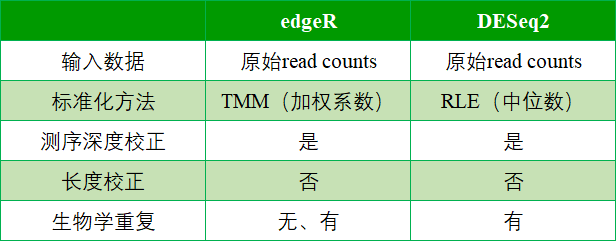
下面我们具体探讨关于这两个软件的比较信息:
①两个软件输入数据均为原始read counts,因为两个软件均是基于负二项分布进行后续分析,若输入RPKM或FPKM可能阻碍软件正确估计数据中的均值和方差的关系。
②两个R包对输入的counts数也都有自己的标准化方法,edgeR——TMM(Trimmed Mean of M-values)和DESeq2——RLE(Relative Log Expression)。无论是edgeR还是DESeq2,都会对测序深度进行校正。如果你对这两种标准化方法比较感兴趣,可查看微信文章 《便利的R包在你进行差异分析的时候帮你做了什么》 。
③这两种软件,均无需调整基因长度。因为开发者认为基因长度对每个RNA样本的读取计数具有相同的影响。
④对于有生物学重复,可利用edgeR和DESeq2进行差异分析。针对无生物学重复,只能利用edgeR开展差异分析。因为edgeR进行差异分析提供了一些备选方案,允许在没有生物学重复的情况下,人为设定离散系数的大小。这个数值可以根据以往项目的经验值进行合理的选择,以便软件可以完成差异检验。
关于差异分析两个软件的选择问题:
目前DESeq2使用频率较高,被引次数32,271,可以说是做差异分析受欢迎的“天花板”;edgeR被引次数23,261,也是比较受欢迎的差异分析工具,大家在分析软件的选择上也不必太过于纠结。一般情况下,若实验有生物学重复,推荐使用DESeq2,无生物学重复只能选用edgeR进行差异分析,edgeR允许没有生物学重复的情况下人为设定离散系数的大小。当然大家如果想做差异分析也可登录OmicShare上传reads数据和分组信息,选择edgeR或DESeq2软件进行分析。如果是我们基迪奥的客户,想选择最优差异分析的结果,可直接登录Omicsmart平台,软件会同时用丰富的图形可视化差异分析的结果。
图4 OmicShare(左)和Omicsmart(右)差异分析软件及参数选择
Q3:在计算差异倍数时,表达量为0的基因是如何处理的?
答:两个软件在进行差异统计检验的时候,reads数为0的基因是允许存在的(不影响检验)。但在计算基因表达差异倍数的时候,如果处理组(分母)的表达量是0,当然就会出现不可除(分母不能为0)。所以,一般情况下会给表达量为0的基因赋予一个极小值(例如0.001),这样既不明显影响结果,又解决了表达差异倍数不可计算的问题。
Q4:如果实验组与对照组相比,reads数相差很大,且实验组表达量为0,这些基因比较重要吗?
答:需要结合统计学结果(P值或FDR值)综合判断这些基因是否属于差异基因。如果这些基因属于差异基因,同时判断处理效应会产生抑制效应,可以重点关注这部分基因。
Q5:差异基因筛选阈值为多少?在两个样品间的差异大小怎么判断?
答:在筛选差异基因过程中,依据经验值通常使用差异倍数(|Fold Change|)≥2且FDR<0.05进行筛选,也是目前发文比较认可的筛选标准。其中,FDR值越小差异越显著,log 2 |FC|越大,差异倍数越大。
Tips
① 转录组差异分析的筛选标准差异倍数≥2,FDR<0.05只是经验值。一般差异倍数可以在1.2~2倍间适当浮动调整,FDR值可以在0.1~0.01之间浮动调整;
② 关于P值和FDR值选择的小问题,一般首选FDR值进行筛选。因为转录组数据中每个基因的差异分析是独立进行,所以差异分析本质上可能进行了上以万次单个基因的差异分析。为了降低假阳性,需要对P值进行矫正,获得矫正后的p值,称为FDR值或Q value。关于多次统计检验后的P值如何矫正,具体的这部分内容解释,可在OmicShare论坛搜索帖子:《显著检验与多重检验校正》。
链接:
https://www.omicshare.com/forum/thread-260-1-1.html
Q6:如果差异分析后获得的差异基因过多或过少,该如何进行调整?
答:我们首先要追其源,了解造成这两大问题常见的原因有哪些:
表2 影响差异基因数量可能的原因
那么围绕这两大核心问题,着手于分析的角度进行讨论,调整方式如下:
差异基因太少:
①若重复性很好,可适当放宽鉴定阈值。如FC>1.5,FDR值换为P值重新筛选 [4];
②若重复性不好,可考虑剔除离群样本(重复数>3),降低组内差异,再重新分析。
差异基因太多:
①若想挖掘差异更显著的基因,可适当将阈值调整的更加严格。如: log2 |FC|>1.5,FDR<0.01 [5];
②进一步做功能富集分析,锁定目标通路类型(如研究与抗病相关通路),再集中解析富集的基因。
今天对大家所关心的差异分析小问题的整理就先分享到这里,如果大家对差异表达分析感兴趣,也可以通过OmicShare在线课堂的对应专题课进行学习,网址给你们留下啦:
https://www.omicshare.com/class/home/index/series?id=2
▼
参考文献
▼
[1] Pimentel, H., Bray, N., Puente, S. et al. Differential analysis of RNA-seq incorporating quantification uncertainty. Nat Methods 14, 687–690 (2017). https://doi.org/10.1038/nmeth.4324
[2] Love, Michael I et al. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology vol . 15,12 (2014): 550. doi:10.1186/s13059-014-0550-8
[3] Robinson, Mark D, et al. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics (Oxford, England) vol. 26,1 (2010): 139-40. doi:10.1093/bioinformatics/btp616
[4] Cui, Qihao et al. “RNA sequencing (RNA-seq) analysis of gene expression provides new insights into hindlimb unloading-induced skeletal muscle atrophy.” Annals of translational medicine vol. 8,23 (2020): 1595. doi:10.21037/atm-20-7400
[5] Hollmén, M., Roudnicky, F., Karaman, S. et al. Characterization of macrophage - cancer cell crosstalk in estrogen receptor positive and triple-negative breast cancer. Sci Rep 5, 9188 (2015). https://doi.org/10.1038/srep09188
- 本文固定链接: https://maimengkong.com/zu/871.html
- 转载请注明: : 萌小白 2022年4月15日 于 卖萌控的博客 发表
- 百度已收录
