宏基因组(Metagenome)和宏转录组(Metatranscriptome)是通过鸟枪法测序技术(Shotgun sequencing),结合全微生物组关联分析(Microbiome-Wide Association Studies,MWAS)的策略,分别从DNA/RNA水平,全面精细地展示整个微生物群落的物种组成谱、功能代谢谱、表达谱,进而从原理上阐明微生物群落在生态系统中发挥作用的根本机制。
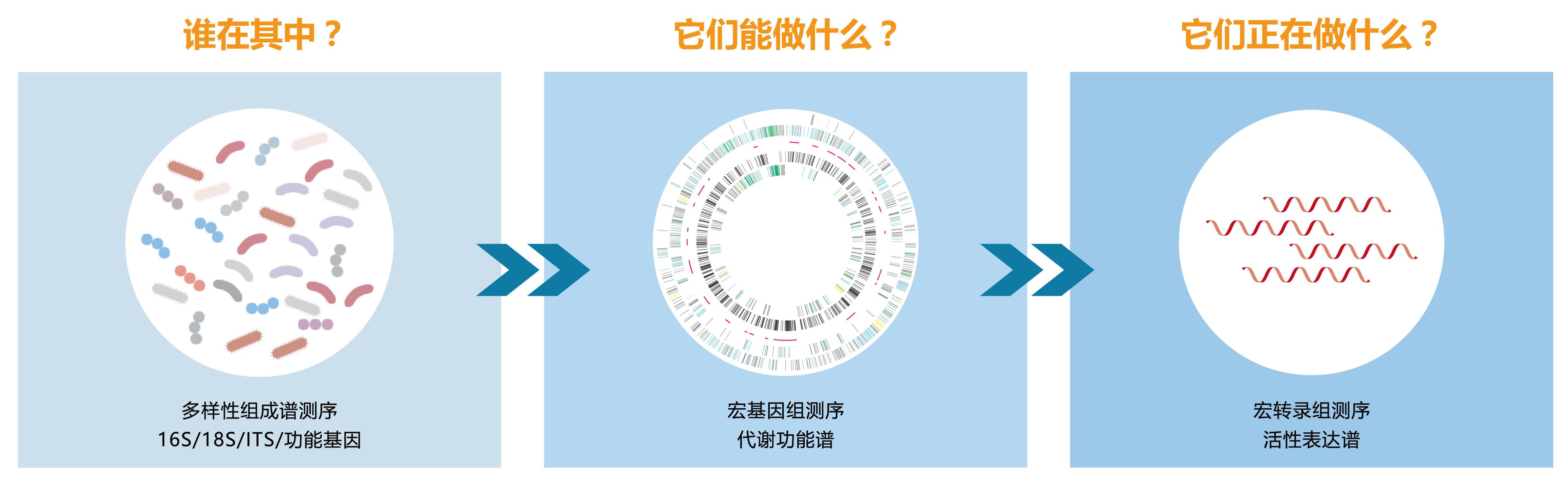
对于数据量和信息量都非常庞大的宏组学研究而言,选对分析软件至关重要。历经多年发展,目前能用于宏基因组和宏转录组分析的软件已然数不胜数,对于分析者而言,可谓“甜蜜的烦恼”!
那么,在琳琅满目的分析方法中,究竟哪些才是当前宏组学的“网红”呢?我们特意花时间整理了以下推荐软件,大家走过路过千万不要错过哦~
★ FastQC ★
FastQC能全方位评估测序产生的原始数据的质量,并通过网页形式展示图形化报告,是测序数据质控的经典工具。
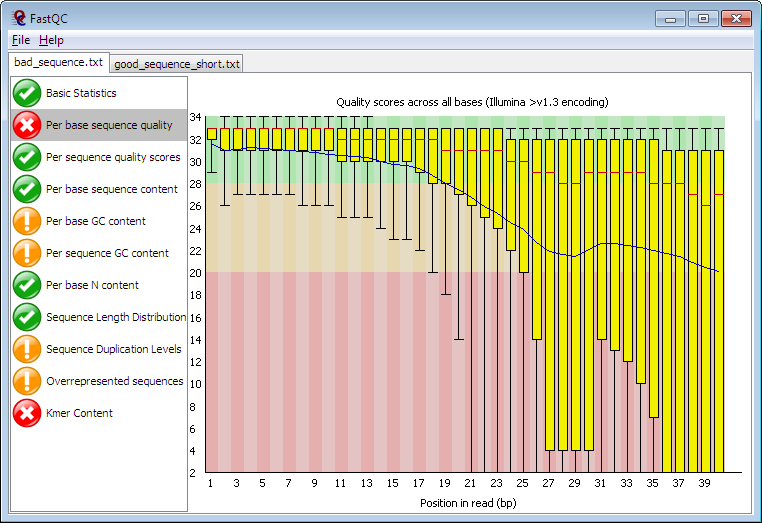
https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
★ fastp ★
fastp是新近发表的测序质控软件,能对数据自动进行全方位质控,并生成人性化的报告。与FastQC相比,fastp运行速度快,功能多,号称“all in one, one for all”。生成的报告中,所有的图表都是使用JavaScript动态绘制,交互功能非常强大。
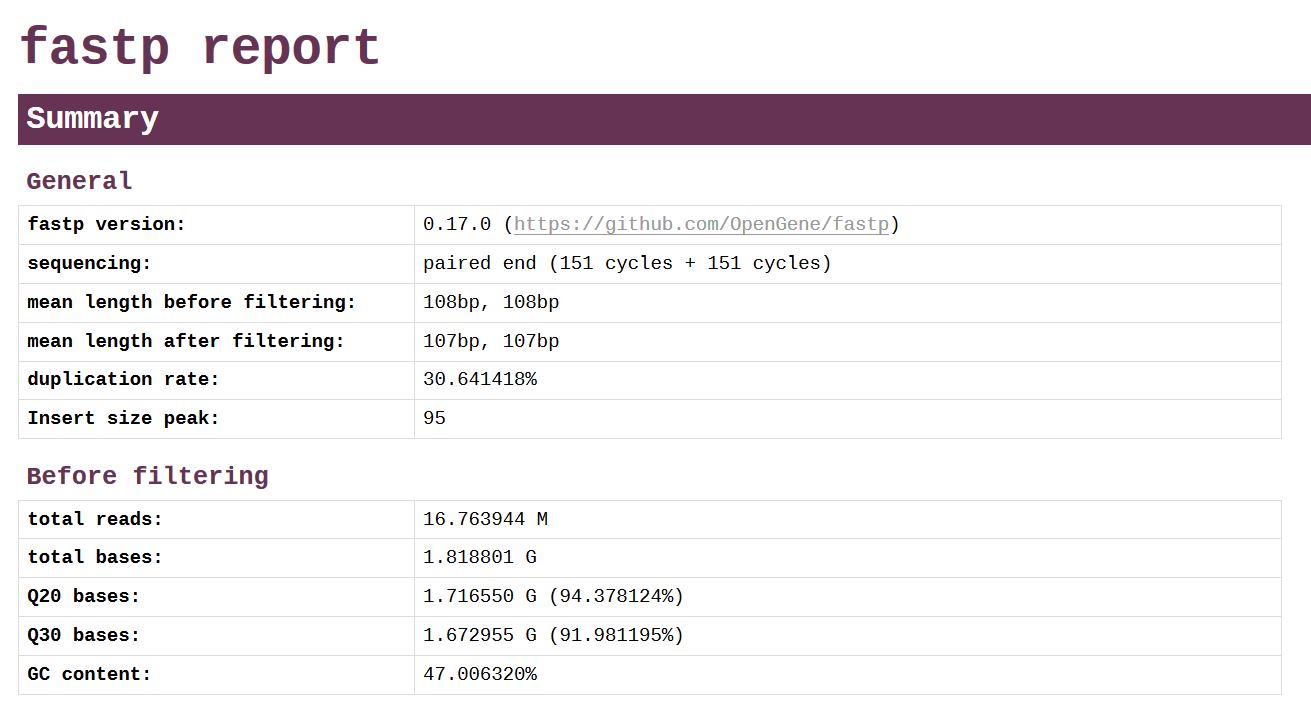
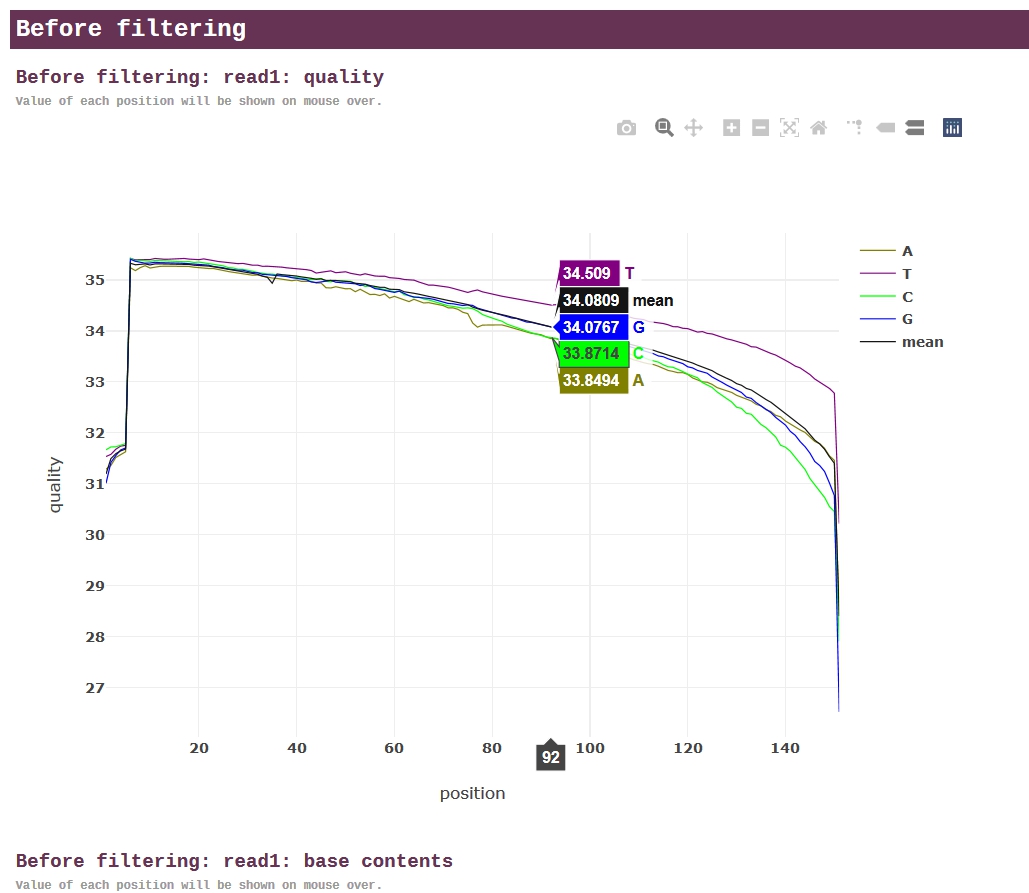
https://github.com/OpenGene/fastp
★ BMTagger ★
对于存在宿主等污染的宏基因组/宏转录组测序数据,可以使用BMTagger软件将质控后的高质量序列与宿主基因组参考序列比对,并舍弃匹配上宿主的序列,以此来尽可能地去除宿主污染序列。

ftp://ftp.ncbi.nlm.nih.gov/pub/agarwala/bmtagger/
★ SortMeRNA ★
对于宏转录组或RNA类的宏病毒组数据,SortMeRNA是去除测序结果中残余的核糖体RNA(rRNA)序列的不二之选。
http://bioinfo.lifl.fr/RNA/sortmerna/
★ Kraken2 ★
Kraken2是Kraken的升级版本,可以通过使用精准k-mer匹配,对非拼接序列实现高精度且快速的物种注释。Kraken2可同时适用于原核与真核微生物的识别,因而具有广泛的应用。
http://ccb.jhu.edu/software/kraken2/
★ MEGAHIT ★
MEGAHIT是一款基于无损压缩的Succinct de Bruijn graphs(SdBG)的快速组装工具,因其具有内存消耗小、计算速度快、低错误率、安装便捷、较长的重叠群长度等优点,在来源较复杂、数据规模较大的宏基因组序列的快速组装上优势明显,是一款应用广泛的主流组装软件。拼接组装时,可以根据样品和数据特性,选择meta-large或meta-sensitive等组装模式。
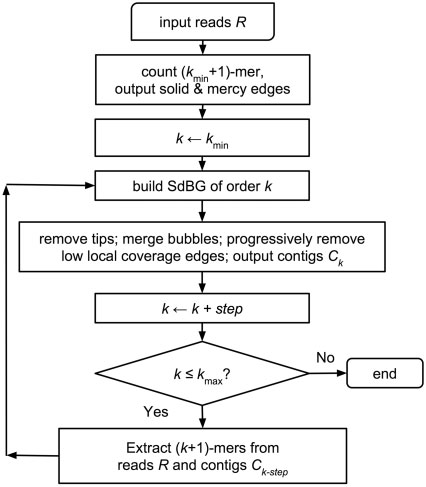
https://hku-bal.github.io/megabox
★ Minimap2 ★
DNA/mRNA序列快速比对神器,相较于BWA-MEM、Bowtie2等常用比对工具比对速度更快,内存占用更少,可适用于二代、三代测序数据。
https://lh3.github.io/minimap2/
★ DIAMOND ★
目前除了BLASTP之外最流行的蛋白比对软件,比对速度快、内存占用少、灵敏度高,可谓集万千优点于一身。

http://github.com/bbuchfink/diamond
★ Blast2lca ★
对于基于拼接序列的物种注释,由于并非所有Contigs序列都具有足够的长度和特异性,它们可能会在注释比对时,同时匹配上多条参考序列,而这些匹配的参考序列又可能分属于不同的物种分类单元;为使分析严谨可靠,同时又不丢失生物学意义,可以采用Blast2lca软件的“最近共同祖先(Lowest Common Ancestor,LCA)”算法,将参考序列分化为不同物种分枝前的最后一级共同分类,作为目标序列的物种分类注释信息。该算法的原理与MEGAN软件相同,是物种注释的主流方法。
https://github.com/emepyc/Blast2lca
★ MetaGeneMark ★
MetaGeneMark是专门用于预测原核微生物和宏基因组基因序列的老牌软件,可以识别组装得到的Contigs序列中的开放阅读框,并预测其中的编码区域,从而获得对应的基因和蛋白序列,运算速度快。
http://exon.gatech.edu/GeneMark/
★ MetaWRAP ★
MetaWRAP是一款整合了质控、拼接、分箱(Binning)、提纯、评估、物种注释、丰度估计、功能注释和可视化的分析流程,纳入超140个工具软件,可一键安装。MetaWRAP流程整合了CONCOCT、MaxBin、MetaBAT等三款分箱工具以及提纯和重组装算法,能提供出色的分箱组装效果,从而有助于从宏基因组数据挖掘单菌基因组信息。
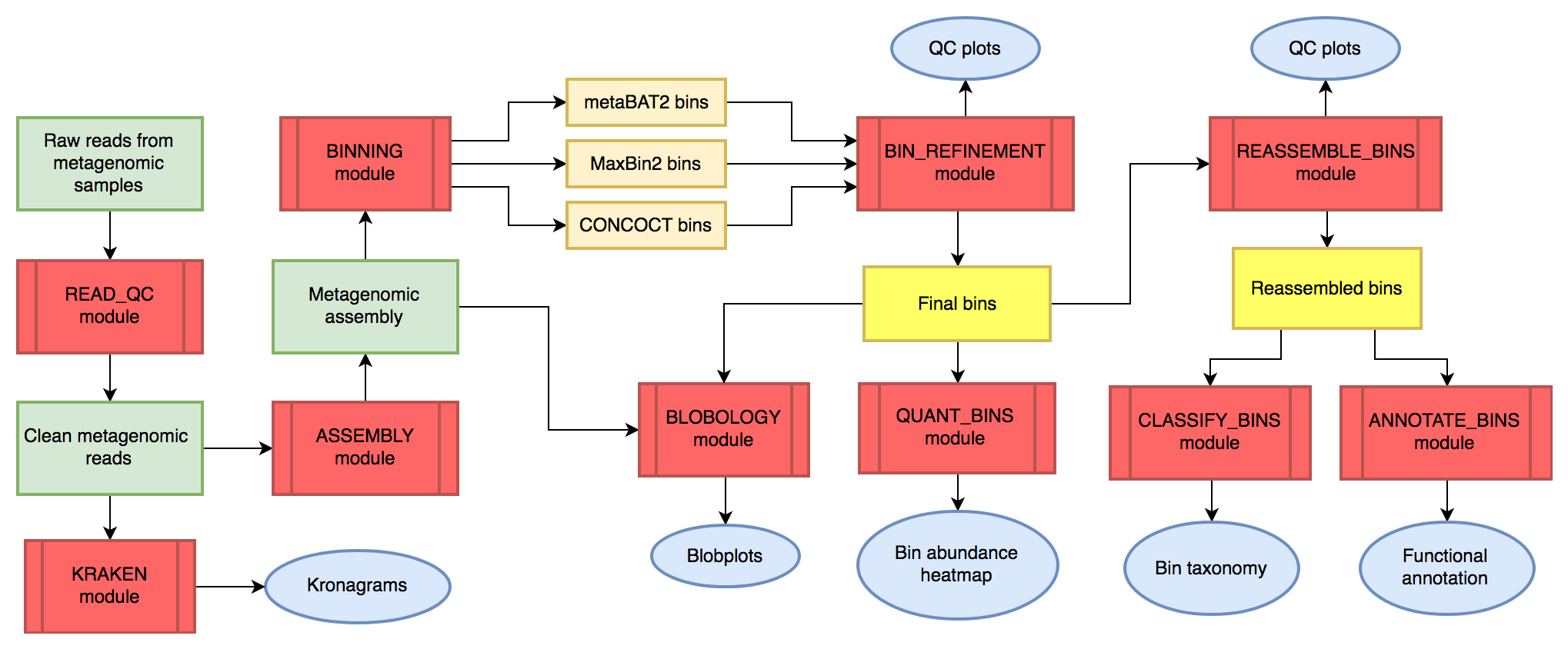
https://github.com/bxlab/metaWRAP
以上就是我们精心整理的宏基因组和宏转录组的常用软件推荐,在此给大家抛砖引玉啦~这些软件,也都已经包含在派森诺最新宏组学分析流程中了哦,欢迎大家尝鲜体验!

- 本文固定链接: https://maimengkong.com/zu/749.html
- 转载请注明: : 萌小白 2021年8月17日 于 卖萌控的博客 发表
- 百度已收录
