在单细胞样本制备过程中,新鲜组织经历解离-离心-重悬-过滤-质控(计数和活率)-上机等步骤。这一过程具有破坏性,根据裂解后的细胞释放出的mRNA反转录成的cDNA进行测序,不可避免地导致 失去了不同细胞之间的空间位置信息和细胞分化情况。单细胞轨迹分析应运而生,轨迹分析可以通过构建细胞间的变化轨迹来重塑细胞随着时间的变化过程,帮助人们从单细胞水平推断细胞之间的演化及分化过程。简单来讲 细胞轨迹分析就是了解不同细胞类型之间的分化关系。目前进行细胞轨迹分析的方法主要有两种,一种是以Monocle软件为代表的 拟时序分析(pseudotime analysis),另一种则是以velocyto/scVelo为主的 RNA速率分析(RNA velocity)。今天,让我们一起学习下单细胞转录组分析之轨迹分析。
1.什么是轨迹分析?
在细胞整个的生命周期中,细胞会从一种功能状态分化到另一种功能状态,处于不同状态的细胞表达不同的基因从而发挥各自的功能,但细胞的瞬时状态很难被描述。由于这个过程是连续发生的,我们可以使用轨迹分析,也叫轨迹推断(trajectory inference),根据不同瞬时状态细胞之间表达模式的相似性,对单细胞沿着轨迹进行排序,模拟细胞动态变化的过程,重建细胞的分化轨迹或者拟时间轴。
由轨迹分析的定义我们可以知道并不是所有的组织都适合轨迹分析。比如骨髓中存在很多干细胞,前体细胞,未成熟细胞到成熟细胞的分化过程,所有骨髓样本就适合做轨迹分析;而如果你的样本比如外周血,其中大多数细胞都是分化成熟的细胞,那就不是很适合做轨迹分析。总之大家做之前可以充分调研,参考一下领域内的高分文献,再决定是否要做轨迹分析。
2.文献中怎么用CNV推断?
1.这是一篇刚发表于BMCmedicine上的卵巢高级别浆液性瘤的ScRNA-seq文章:
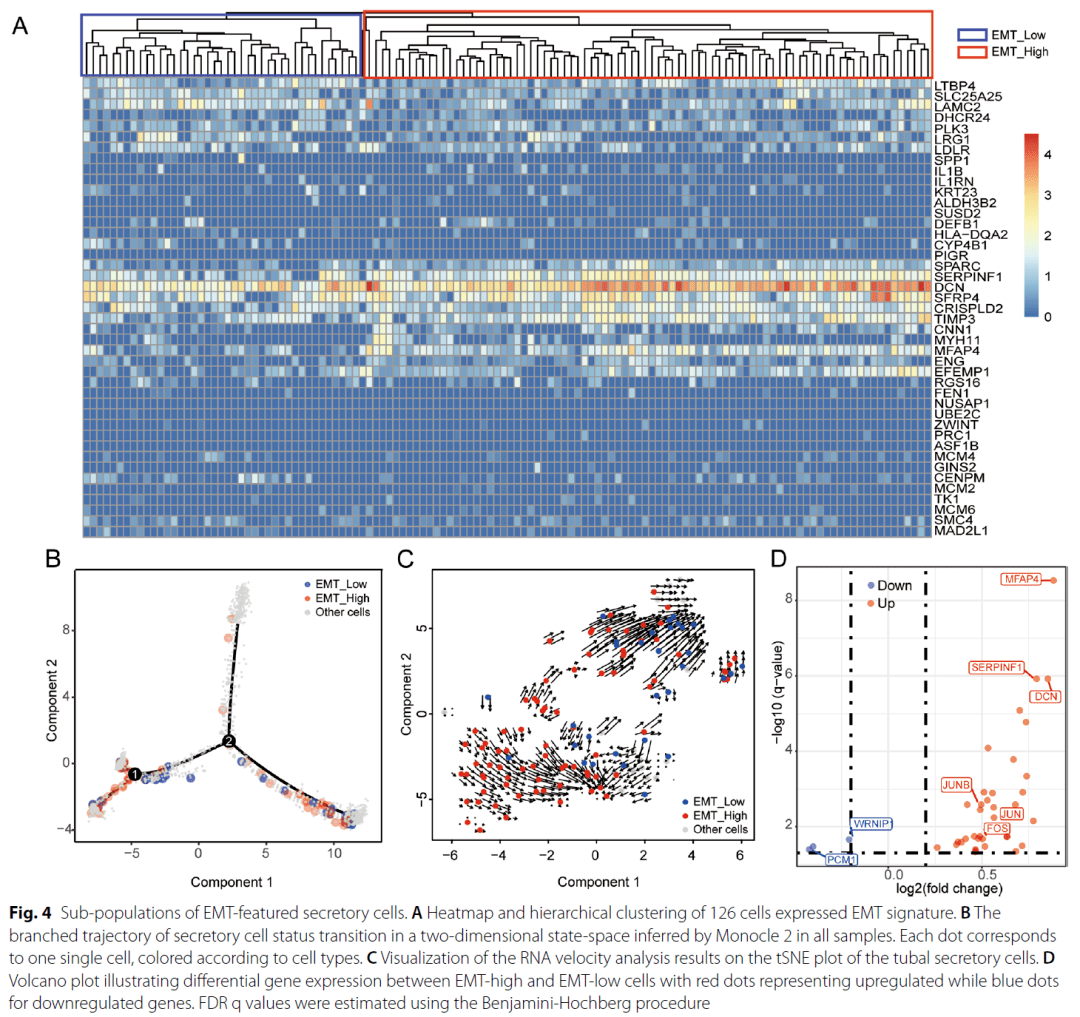
本文通过3例BRCA1突变样本 VS 3例正常对照样本发现了卵巢高级别浆液性瘤的EMT样分泌细胞亚群和免疫逃逸表型。
上图揭示了EMT样分泌细胞亚群的特征:( A) 126个细胞中EMT signature的层次聚类热图。( B) 根据Monocle 2推断所有样本在二维空间中分泌细胞状态转变的分支轨迹。每个点对应一个细胞,根据细胞类型着色。( C) 输卵管分泌细胞的RNA速率的分析结果在tSNE图上的可视化。( D) Volcano图显示了EMT 高和EMT低细胞之间的差异基因表达。
2.这是一篇刚发表于NPGprecision oncology上的晚期胆道癌ScRNA-seq景观的文章:
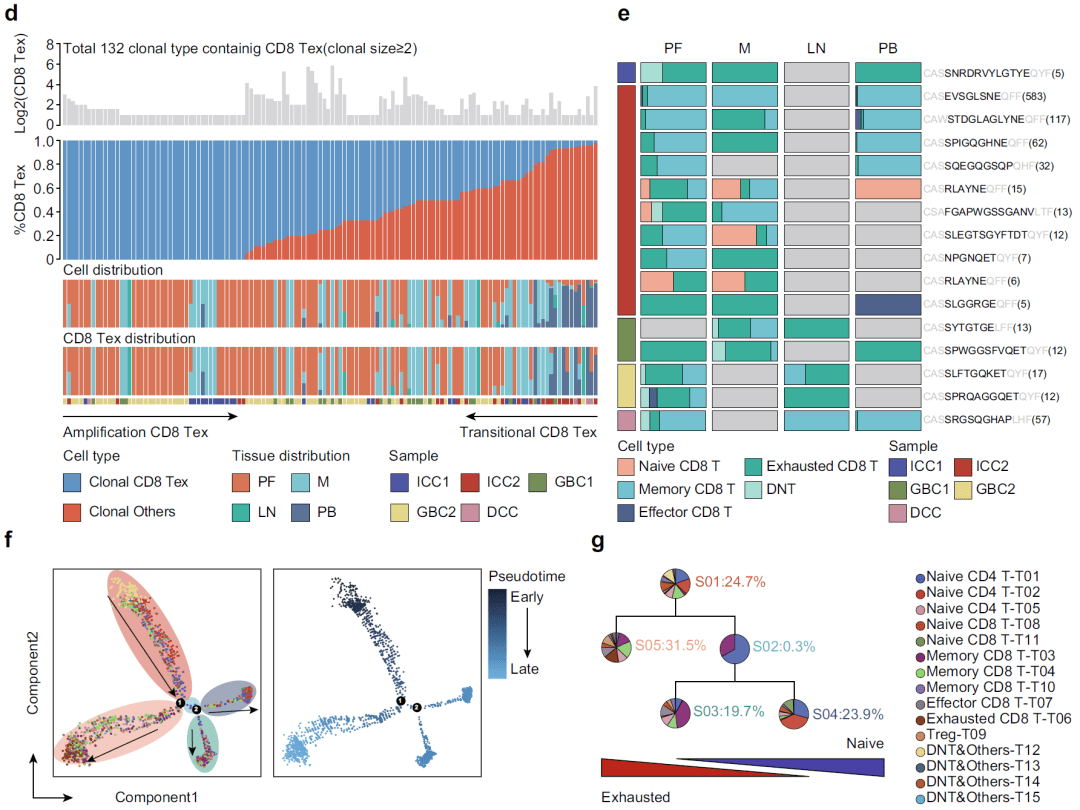
为了全面表征晚期胆道癌的免疫微环境,作者对5个病人的原发灶、转移灶和外周血进行ScRNA-seq测序。结合TCR,发现耗竭性的CD8+T细胞在TME中保持克隆扩增和高度增殖。( D) 患者不同组织中耗竭性CD8+T细胞的克隆类型,从上到下显示每个克隆类型对应的耗竭CD8+T细胞数量、耗竭CD8+T细胞和移行CD8+T细胞之间的比值、患者不同组织中的细胞和耗竭CD8+T细胞分布。( E) 具有相同TCR β等位基因的不同组织中不同 T 细胞亚型的分布。( F) 细胞亚群和伪时序注释的T细胞的Monocle 2轨迹。( G) 伪时序轨迹中不同状态下T 细胞组成分数的饼图。
3.该如何进行时序分析?
这里着重介绍常用的 拟时序分析和 RNA速率。
1.拟时序分析
为了解决单细胞测序导致的不同细胞类型之间分化关系不明的问题,在2014年的一篇Nat Biotechnol文章中,Cole Trapnell等提出了拟时(pseudotime)的概念:即以细胞的分化状态来模拟时间轴,具有高度分化潜能的细胞拥有更初始的时间,分化程度较高的细胞时间则相对靠后。基于此,一个样本中包含了各个亚型中处于不同分化进程的细胞,按照分化潜能这些细胞分别对应了时间轴中的不同时间,我们可以据此模拟出分化轨迹。因为样本中的细胞实际上是在同一个时间点获得的,因此这个时间被称为拟时(pseudotime)。21年底微信有一篇推文题目是: 拟时序分析就是差异分析的细节剖析,大家可以根据拟时序的原理试着理解下。接下来,小王子分别阐述 Monocle, Monocle 2, 和 Monocle 3的演进:
(1) Monocle: Thedynamics and regulators of cell fate decisions are revealed by pseudotemporalordering of single cells. Nat Biotechnol. 2014 Apr. PMID: 24658644.
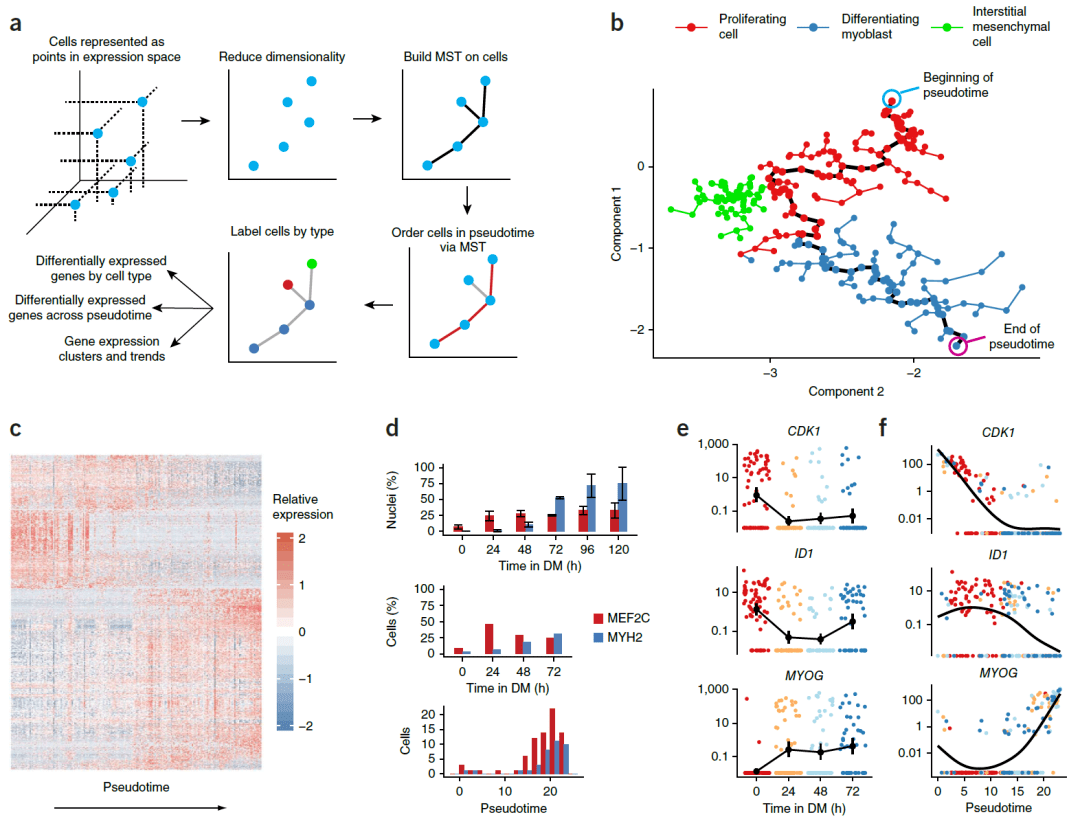
Monocle 通过分化的进程对单个细胞进行排序
(a) Monocle算法概述。
(b) 二维独立成分空间中的细胞表达谱(点)。连接点的线代表 Monocle 构建的最小生成树(MST)的边缘。黑色实线表示MST的主要直径路径,并提供了单元格伪时间排序的骨架。MST只有轨迹,没有起点和终点,需要先验知识。
(c) 通过Monocle(行)鉴定的差异表达基因的表达,细胞(列)以伪时间顺序显示。排除间充质细胞。
(d) 条形图显示收集时免疫荧光测定的 MEF2C 和 MYH2 表达细胞的比例(上),收集时的RNA-Seq(中)或伪时RNA-Seq(下)。
(e) 肌肉分化的关键调节因子的表达,按收集的时间排序。
(f) e中的监管者,由 Monocle 在伪时间中排序。e、f中的点按采集时间着色。
(2) Monocle 2:Reversedgraph embedding resolves complex single-cell trajectories. Nat Methods. 2017Oct. PMID: 28825705.
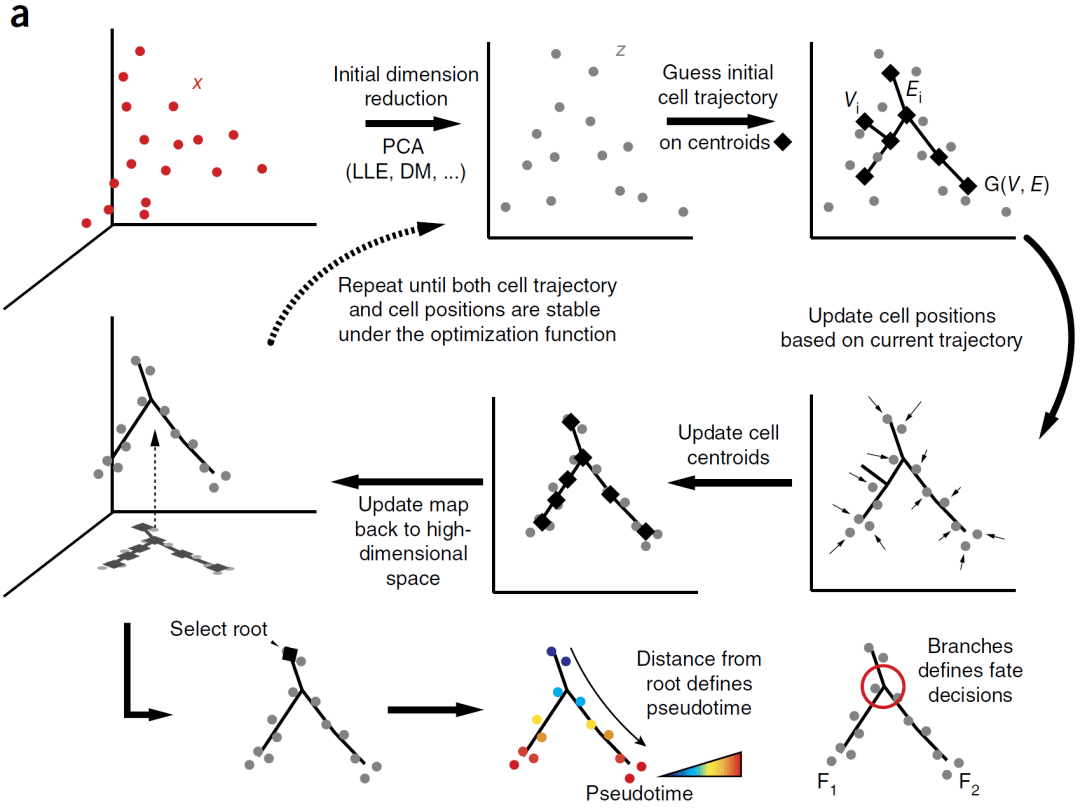
Monocle 2是做单细胞拟时序最常用的R包
主要改进点:定义轨迹时利用的是反向图嵌入(RGE),而Monocle用的MST。
MST:高度依赖每个点的位置以及点与点之间的距离,假设某个点的位置有些微变化就会得到完全不同的细胞轨迹。
RGE:先对细胞进行聚类,再对细胞群的平均值进行轨迹构建。
(a)Monocle 2通过RGE自动学习单细胞轨迹和分支点。每个细胞在高维空间中表示为一个点(x) ,其中每个维度对应于排序基因的表达水平。
通过PCA等降维方法将数据投射到低维空间 (z) 上,Monocle 2在使用k-means聚类自动选择的一组质心(菱形)上构建生成树。
然后将细胞向最近的树顶点移动,顶点位置更新为“拟合”细胞,之后学习新的生成树,并迭代该过程,直到树和细胞收敛。
然后,用户选择一个尖端作为“根”(根据先验知识),每个细胞的伪时间计算为其沿树到根的距离,并根据主图自动分配其分支。
(3) Monocle 3:Thesingle-cell tranional landscape of mammalian organogenesis. Nature. 2019Feb. PMID: 30787437.

Monocle 3还在不断更新中,其原理和流程与Monocle 2类似,降维—聚类—拟时间轴的建立—差异分析,改动还是比较大的,其内置大部分的函数名称都改变了
#代码实战:目前还是Monocle使用的最多,网上教程也非常多, #这里给大家放一个最常规的,详细内容可根据文后的参考大家在网上了解。 #使用TISCH中的Seurat(LIHC的GSE125449)数据----------------------------------------------------------------------- library(Seurat) library(monocle) library(DDRTree) library(ggsci) library(dplyr) library(ggplot2) library(export)#获得Count矩阵Seurat <- Read10X_h5('LIHC_GSE125449_aPDL1aCTLA4_expression.h5')Seurat <- CreateSeuratObject(Seurat, project = "", min.cells = 3, min.features = 200)Seurat #3835个细胞,17356个基因gc
meta <- read.delim("LIHC_GSE125449_aPDL1aCTLA4_CellMetainfo_table.tsv", header=F)colnames(meta) <- meta[1,]meta <- meta[-1,]rownames(data)colnames(meta)colnames(meta) <- c("Cell","UMAP_1","UMAP_2","Celltype_malignancy", "Celltype_major_lineage","Celltype_minor_lineage","Cluster","Celltype","Treatment","Patient","Sample","Source","Age","Gender","Stage")
Seurat@assays$RNA@meta.features <- as.data.frame(rownames(Seurat))identical(meta$Cell,colnames(Seurat))meta <- meta[meta$Cell%in%colnames(Seurat),]
a <- Seurat@meta.dataa <- cbind(a,meta)Seurat@meta.data<- atable(Seurat@meta.data$Cluster)
#数据质控+去除批次效应 ---------------------------------------------------------Seurat[["percent.mt"]] <- PercentageFeatureSet(Seurat, pattern = "^MT-") # 注意基因名称变化VlnPlot(Seurat, group.by= "Patient", features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), pt.size = 0, ncol = 3) + NoLegend
##筛选合格细胞cat("Before filter:",nrow(Seurat@meta.data),"cells\n")Seurat <- subset(Seurat, subset = nFeature_RNA > 200& nFeature_RNA < 3000& nCount_RNA > 500& nCount_RNA < 4000&percent.mt < 5)cat("After filter :",nrow(Seurat@meta.data),"cells\n")Seurat
monocle.matrix <- as.matrix(Seurat@assays$RNA@data)monocle.sample <- Seurat@meta.datamonocle.geneAnn <- data.frame(gene_short_name = row.names(monocle.matrix), row.names = row.names(monocle.matrix))monocle.sample$Cluster <- monocle.sample$Cluster
data<- as(as.matrix(monocle.matrix), 'sparseMatrix')pd<-new("AnnotatedDataFrame", data= monocle.sample)fd<-new("AnnotatedDataFrame", data= monocle.geneAnn)cds <- newCellDataSet(data, phenoData = pd, featureData = fd)gccds <- estimateSizeFactors(cds)cds <- estimateDispersions(cds)cds <- detectGenes(cds, min_expr = 0.1)print(head(fData(cds)))
Idents(Seurat)="Cluster"Seurat.markers <- FindAllMarkers(object= Seurat,only.pos = FALSE,min.pct = 0.25,logfc.threshold = 1)sig.cellMarkers <- Seurat.markers[(abs(as.numeric(as.vector(Seurat.markers$avg_log2FC)))>1& as.numeric(as.vector(Seurat.markers$p_val_adj))<0.05),]
gctable(Seurat.markers$cluster)
sig.cellMarker <- Seurat.markers %>% mutate(Ratio = round(pct.1/pct.2,5),pct.fc = pct.1-pct.2) %>%filter(as.numeric(as.vector(p_val_adj))<0.001) # 本条件为过滤统计学不显著的基因
table(sig.cellMarker$cluster)
rm(a,data,fd,meta,monocle.geneAnn,monocle.matrix,monocle.sample,pd,Seurat)gc
cds <- setOrderingFilter(cds,ordering_genes = sig.cellMarker$gene) ##设置ordercds <- reduceDimension(cds, max_components = 2, method="DDRTress")gc
cds <- orderCells(cds)gc
cols <- c(pal_lancet(9)[c(1:9)],pal_jco(10)[c(1:10)])plot_cell_trajectory(cds,show_branch_points=F,color_by = "Cluster", cell_size = 2)+scale_colour_manual(values = cols[c(1:15)])+theme(axis.title = element_text(size=18),axis.text = element_text(size=18,colour = 'black'),legend.title = element_text(size=36),legend.text = element_text(size=36),legend.position = c(0.8,0.8))+guides(color = guide_legend(nrow = 3))graph2pdf(file="Monocle1.pdf",width=11,height=8)
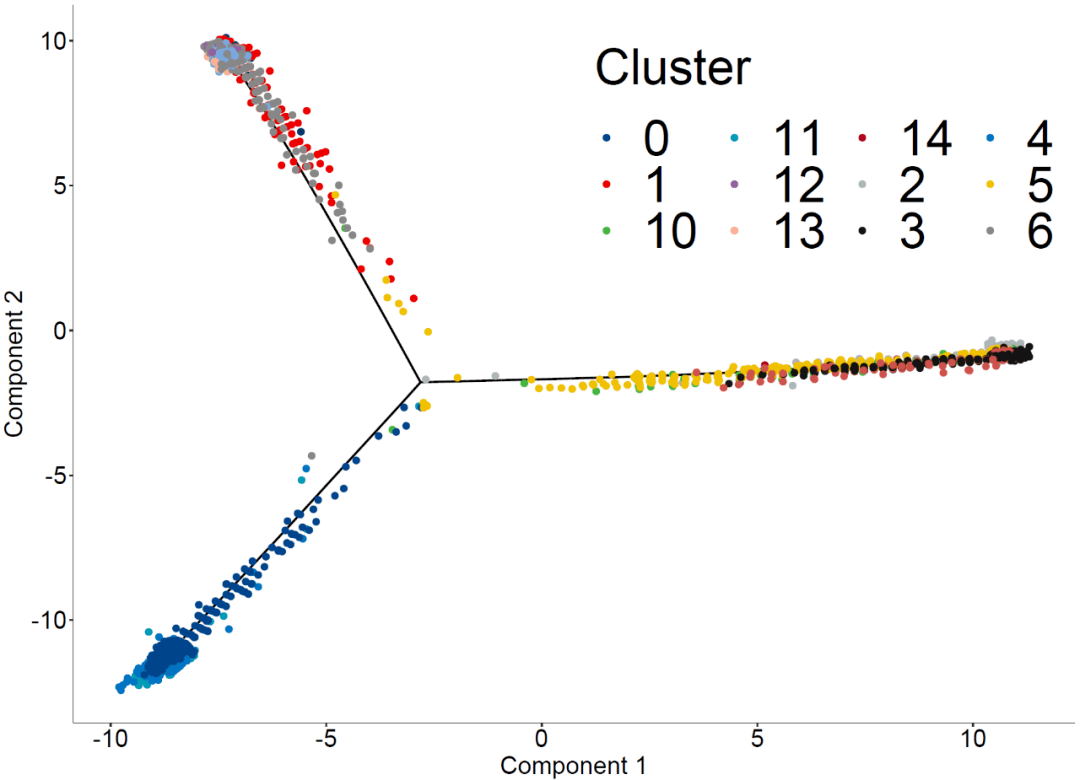
温馨提示:如果你跑cds <- orderCells(cds)时出现了以下问题,各种Google的解决方案都不行时,这里建议把R4.2.1的版本切换成较低版本比如R4.0.5
2.RNA velocity
轨迹分析的核心在于根据瞬态细胞之间的表达模式相似性,对细胞沿着轨迹进行排序,重建不同亚群之间的分化轨迹和伪时间轴,依此来模拟细胞动态变化的过程。
RNA丰度是衡量细胞状态的有力指标。ScRNA-seq可以以准确度、灵敏度和高通量地揭示 RNA 丰度。然而,由于取材的瞬时性,这种方法仅捕获某个时间点的静态快照。这里,作者介绍了RNA速率--基因表达状态的时间导数--可以通过区分普通ScRNA-seq数据中未剪接和剪接的mRNA来直接估计。RNA速率是一个高维向量,可在数小时的时间尺度上预测单个细胞的未来状态。
RNA速率分析的前提是我们要从ScRNA-seq数据中区分出未剪接(未成熟)和剪接(成熟)的mRNA,分析时需要从fastq文件开始,与基因组比对后提取剪接、未剪接、不确定的信息。
4.RNA velocity:RNAvelocity of single cells. Nature. 2018 Aug. PMID: 30089906.
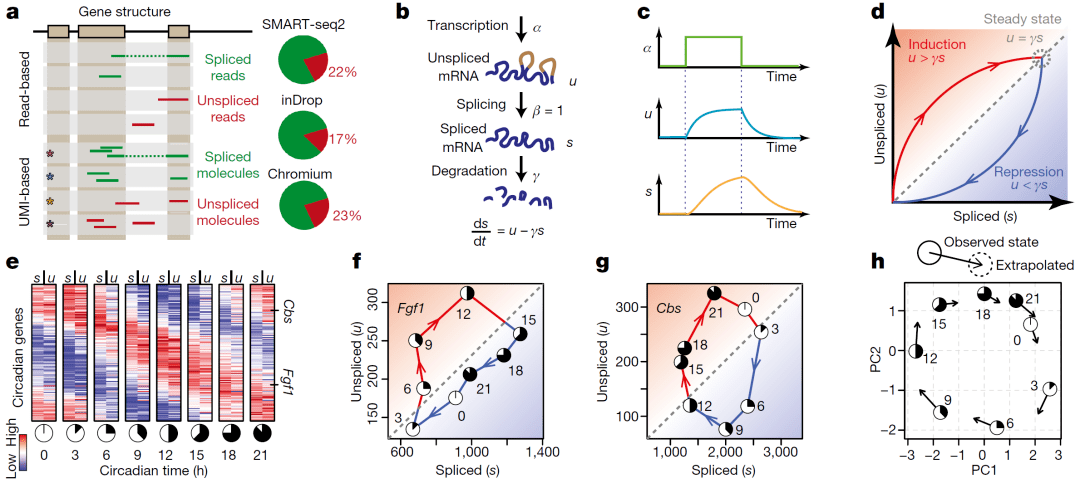
未剪接和剪接mRNA之间的平衡可预测细胞状态进展
(b) 转录动力学模型,捕获参与产生未剪接 (u) 和剪接 (s)mRNA产物的转录(α)、剪接(β) 和降解(γ) 速率。
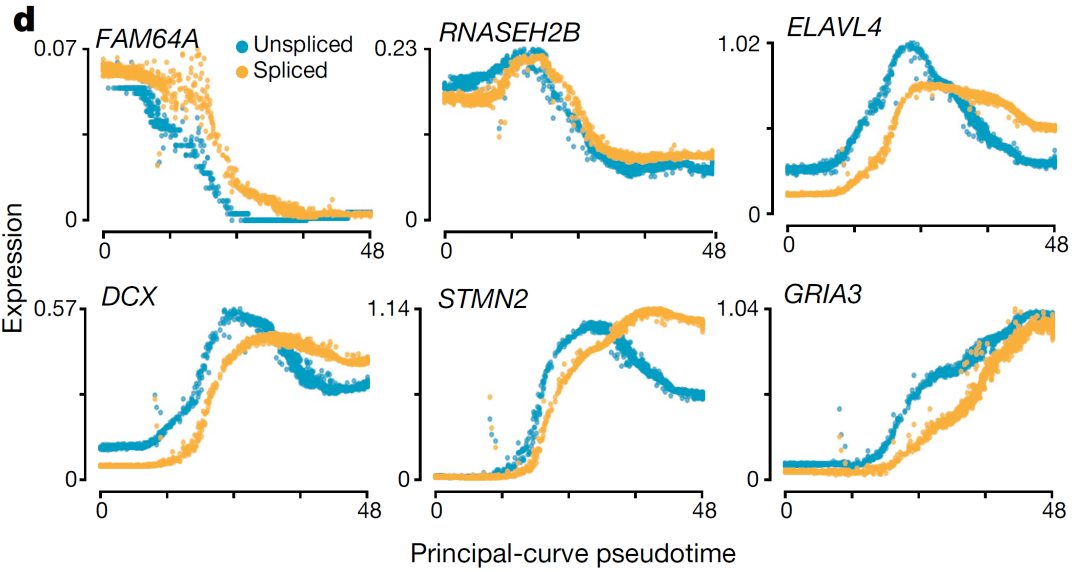
(d) 6个示例基因的谷氨酸能神经元成熟过程中的伪时间表达谱。未剪接的细胞出现早于已剪接的。
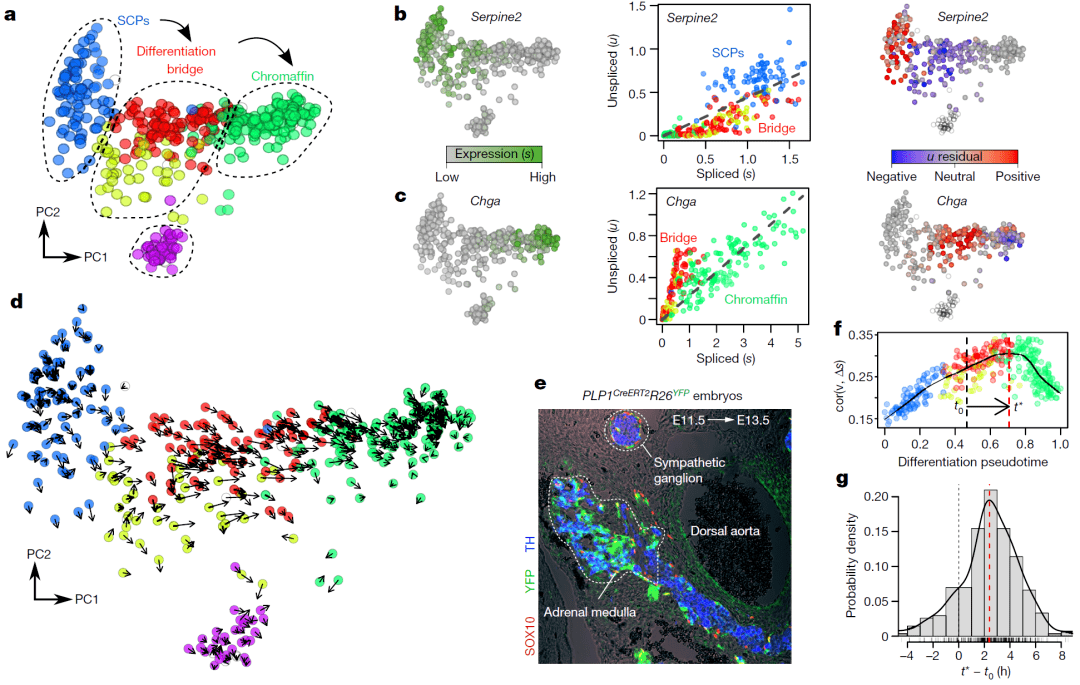
RNA速率概括了嗜铬细胞分化的动力学
(a) PCA投影显示胚胎12.5日龄小鼠(n = 385个细胞)雪旺细胞前体 (SCPs) 分化为嗜铬细胞的主要亚群。
(b-c) 表达模式(左),未剪接-剪接相位图(中心,根据 a 对细胞进行着色),显示了被抑制的 Serpine2(b) 和诱导的 Chga(c) 基因的 u 残差(右)。合并5个最近细胞邻居的读取计数。
(d) 观察到的和外推的未来状态(箭头)显示在前两个PC 上。在没有细胞或基因合并的情况下估计RNA速率。
与Monocle系列的拟时序分析的主要区别:RNA 速率根据mRNA有无剪接推断(从未剪接mRNA到剪接mRNA分化),可以在无先验知识的前提下预测谱系的方向。
小结
总的来说,单细胞的拟时序分析就是通过不同瞬时状态细胞之间表达模式的相似性,对单细胞沿着轨迹进行排序,模拟细胞动态变化的过程,重建细胞的分化轨迹或者拟时间轴。而RNA速率是从fastq文件开始,从ScRNA-seq数据中区分出未剪接(未成熟)和剪接(成熟)的mRNA(从未剪接mRNA到剪接mRNA分化),可以在无先验知识的前提下预测谱系的方向。时序分析是继细胞通讯、CNV推断外经常使用的单细胞下游分析,感兴趣的同学可以现学现用,立刻就能增加到你的研究中来!
本文主要参考:
1.基迪奥生物-10X单细胞定制分析--细胞分化轨迹分析(上)
2.CSDN博客-小L的生信笔记-细胞轨迹(trajectory)分析——Monocle
3.简书-单细胞系列课程-10 Trajectory inference analysis of scRNA-seq data
4.简书-单细胞之轨迹分析-1:RNA velocity
- 本文固定链接: https://maimengkong.com/zu/1534.html
- 转载请注明: : 萌小白 2023年5月13日 于 卖萌控的博客 发表
- 百度已收录
