全基因组关联分析(genome wide association study, GWAS) 是利用全基因组范围内筛选出的高密度分子标记对所研究的群体进行扫描, 使用扫描得到的分子标记数据与表型性状之间进行关联分析的方法。
GWAS的出现为全面、系统地研究基因组学掀开了新的一页, 在医学中已经利用该方法鉴定出大量与人类复杂疾病或数量性状相关的遗传变异, 成为研究人类基因组学的关键手段。而在植物基因组中GWAS也取得了良好的应用效果, 应用GWAS发掘 植物复杂数量性状基因、为植物分子育种提供依据已成为植物基因组学研究的热点。
GWAS定位优势
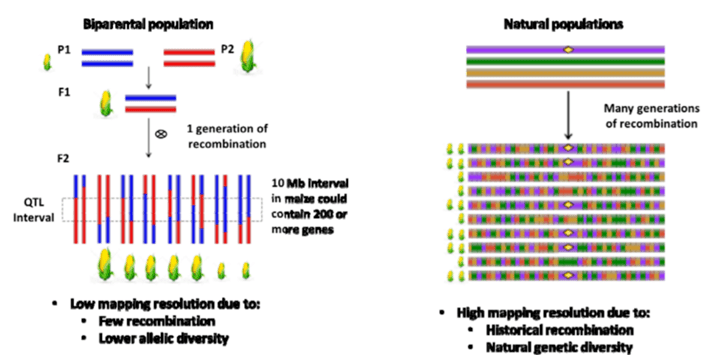
在动植物研究中,GWAS相对于传统的利用遗传图谱做QTL定位有很多优势:
-
节约时间:无需构建遗传群体,GWAS自然群体或者种质资源可以直接用于关联分析;
-
定位精确:人工群体由于群体大小限制,重组不充分,定位区间往往很大,而GWAS使用的自然群体大都经过长时间、多世代的繁衍,基因组重组充分,关联定位准确;
-
GWAS中表型性状变异丰富,只要群体足够大,几乎可以涵盖所有的表型性状,因此GWAS可以关联任何性状。而人工群体由于群体遗传背景单一,如果父母本中某些表型不存在就无法定位该性状。
GWAS-顶级期刊常客
案例1:GB -苦荞-2021群体+gwas文章

基础数据 :510份苦荞种质,包括 32份野生种质、 478份地方种质和 7份荞麦属种质。 本研究对全球 14个国家的 510份苦荞种质资源进行了测序分析,共生成原始数据 3.98 Tb,平均测序深度为 12.65X,基因组覆盖率为 91.72%。最终鉴定了 1,095,748个 SNP和 116,516个 InDel。
案例2:NC –梨-2021群体gwas文章

基础数据 :312个沙梨( Pyrus pyrifolia)品系进行了测序,其中 231个品系是本地品种( landrace), 81个品系是改进的栽培种( improved cultivar)。总测序量产出为 2.15Tb数据,经过比对参考基因组、变异检测以及变异过滤之后得到 340万个 SNP(单核苷酸突变)用于下游研究。(值得注意的是,本研究中使用的参考基因组是白梨 Pyrus bretschneideri,数据与参考基因组的平均比对率是 73.5%,这在涉及不同品系和品种的重测序研究当中,是一个可以接受的数字。)在系统发育与群体结构的分析当中,显示出选择的梨品系大致区分成两类(中国品系 Group I和日韩品系 Group II)。
案例3:NC –毛竹-2021群体gwas文章

基础数据 :毛竹( Phyllostachys edulis)是世界上最重要的竹种,在我国主要分布于华南地区。本研究中,作者对来自 15个代表性地理区域的 427份毛竹进行全基因组重测序,构建了毛竹基因组变异图谱,进行了种群进化分析,并对 9个重要性状进行全基因组关联分析以确定候选基因,揭示了这一无性繁殖物种的群体多样性,为了解毛竹进化和农业重要性状的遗传机制提供了基础与资源。 427份来自于 15个代表性区域的毛竹种质与近缘种 Phyllostachys kwangsiensis, Illumina测序(平均 20X)获得 16.60TB数据。
- 本文固定链接: https://maimengkong.com/zu/1493.html
- 转载请注明: : 萌小白 2023年4月29日 于 卖萌控的博客 发表
- 百度已收录
